IdrD domain-level metagenome surveys
Overview
This is a reproducible, narrative description of the methods used to investigate the representation of idrD in real-world environments, specifically the human microbiome, as reported in our manuscript. The motivation for mining metagenomes came from the biochemical characterization of IdrD, which represents a novel contact-dependent toxin involved in self/non-self identification between bacterial strains. IdrD is particularly interesting, structurally, it contains multiple subregions with varying nucleotide-level conservation. The 5’ end of the gene (N-terminus) seems to behave “ordinarily” with expected nucleotide-level divergence between taxa; the middle usually has a relatively conserved domain for recombination (e.g., Rhs); and the C-terminal domain (CTD) is much more variable. This CTD contains the active site for DNase activity. So, a priori we expect some lumpy coverage reflecting the different domains, as they represent different levels of sequence conservation (i.e., frequency in the pool of short reads that is a metagenome). Thus, we were interested in investigating its presence and diversity in complex, host-associated microbiomes to understand the CTD’s prevalence, diversity, and population specificity.
The process
We started with a FASTA file of full-length idrD nucleotide sequences, obtained by manual curation of idrD hits from a combination of HMM and tBLASTn searches of EMBL and NCBI. The sequences can be found here:
>Acinetobacter_baumannii_strain_XH858_idrD
ATGAACAAGAATAGTTATCGCATTATTTATAGCAAAGCACGTCAAATGTTTGTGGCCGTGGCTGAAAATGTACGAAGCCAAACCAAGACTTCTGGTCAAAGTGAAGCAAGTACCCAGAACAAGATTGATAACACTGAATCTCAGGCATTTCATCAACTCTGGCAAGTGAAAGCATTGGTTGCCAGTATTAGCCTGTGGATGCCTTTAGCACCTGTTTATGCGGGTATTGTAGCGGACAGTGCCGCAAATGCTGCGAATAGAGCAGTTATTGGTGCTGGAAAGAACTCAGCAGGTACAGTCGTTCCTGTCGTTAATATTCAAACTCCAAAAAATGGGATATCCCATAATATTTATAAACAGTTTGATGTATTGGCTGAAGGTGCAGTACTCAATAACAGCCGTCAGGGTGCTACGACAAAAACAGTAGGAAACGTTGCAGCGAACCCATTTTTAGCTACAGGTGAAGCACGAGTTATTTTAAATGAAGTAAATTCATCAGCCGCTTCACGCTTTGAGGGGAACTTGGAAGTTGCAGGGCAAATGGCTGATGTGATTATTGCCAACCCGTCTGGTATTAGTATTAAAGGTGGTGGCTTTATTAATGCCAACAAGGCAATTTTCACCACAGGTAAACCTCAGTTAAATGCTGATGGCAGTATCAAGCAGTTTACGGTTGACCAAGGAAAAATTACGGTCTCAGCGAACCCCAATTCTAAATTTGGTTTGGGTGGAAATAATAACGATGCAAATTATGTGGATCTCTATGCGCGTGCATTAGAGCTTAGTGCTGAGCTACGTGCTAAAAATGATATTCAGGTGATTGCCGGGGCAAATAATGTCAGTGCAGATTTACAGGATGTTACAGCTAAAACAGGTACTGGAACAGCACCAACATTGGCTGTGGATGTTAAAGCATTAGGCGGGATGTATGCCAATAATATTTATTTGATGGGAACTGAAAAAGGCTTAGGCGTGACGAACGCAGGGACTATTCAGGCAGTCAACAATCTGGTAATTACCAGTGCCGGAAAGATTGAACACAGTGGTACAATCAGTTCGACCAGTAAAACTCAAGGTTTAGTAAACATACAAACAACGGGTACGGGTGCAGCTTCCGATATCAACTCATCAGGTAGTATTAATAGTAATAGCATGTTAAATATCGATTCAGGTAATAACCTGAATGTAAATGCTAAAGAAATTATTATTAATAACAGCAGTCTAGCATCTTCACCTTTAATCGTTAATACCAAAGGTAATATCAATTTAGCAGCAAACACCCGCATTATGGATGATTCACAAGGTGGAGATGTCTATATTGATGCAGCAAATATCAATCTTGCCGCTGGATCGGAACTCAAAAGTAACCGTGGTACAGCAACCATACAAGTACAAAAAGACTTAGTTGCAGCCAAAGGCGCAAAACTGATTGCTGCCCAAGACTTAAATGTTTTAAGTAATGGTAAGTTATCACTTACTGAGAACCATATACAGGCATCATTAGGTACAATCAACCTACAAGCAAATTCGGCTAATACCCAAAATCTGATTGACCTTCAAGGTGGAACGATTTATGCGGGTAAAGATTTAAATTTATATAGCACTGATAATATTAATATGAAAAACTTAAGTTTTTCAATAGAAAATAATAAAAATCGAGTAAAAAATATTAATATTTATAGTGGTGGTAGTTTATATTGGGATAATACTGCTTTTTTAATACCTGACCTAACTGGAAAAATTTCACTGAGAGCGGAAAAAGATTTAACAATATTGGGTCAAGGTGGTTTAAGTGCAGATAACGGTATTAGTTTATATGGTGGAAATGTAAATGTTTCTTCACTTAATTTAAATTCAAATATTGGTAATACATTGGTTTTTGCAAATAATAATATTGATATTAAGGGGGTAAATATAAAGAGTAATAATGGAAATTTAAATTTTTCAGCAGAAAATGGCTATATCAAGATTGCTGATTCGAATTTGTCAAGCGAAAAAAATAAATTATTTTTAATTTCTAAAGATAAGCAAATTTTTAATAATACTAGTTTACTTTCAGGAGGGGATTTAATATTAAATACTATAAGTAATAAAGGTGATGTAGAAATTAATCAAGATGGAGGGTTATTTTCTTCACCAATTAAATCAAATAATGGTAATGTAATTATTGATTCTAAAGGGATGTTAAATATAACAACAAAAGACTTAAATAAATTTGTTGATATCAGTGGTGAGAAAATAGAAATTTCTTCTTCTAATAATATGAAATTAGATGGTATTAATGTATATTCAAATGGTAATTTATCTCTACATGCAGATAAAGATATATACTTGAATACAGTTCTGCCAAACTCATGGACAGGGGGTGGTTTTGGTAGTGAAATCATTAGAAGCCAAAAGCATATATCTATCAGTAGTAAAGAGGGCAGTGTTAAAATAGGATTAGAAAACCAAGCTTTAAATGCTCCTGTCGTTGGCCTAAAAGCTAATGGCGTAATTTCAATAAAAAGTAAGTTAGATCAAAGTTATCAAAATACCTCGCTAAATGCGGGTGCTATTACGCTTCACTCAGATGCCGGAAGTATTAATAATAGTAAAAATTTTAGAGCAATTGCTAATACAACATTTTTTCTCGAAAGTGATAGCGAATTAAAAAAAATAAATGGTAACTTAAGTTTATATGCGAAAAAAGATATTCAACTAGACGCGATTAACTCTCAAGGACGAGCTTTTGATCCAAATTACACGCGTATGATTAAACCCGTGCTCTCAAGCCAAGGTTTAATGGATATTCGTGCTGATGGAGTGTTTCAGGTGAATGGTAAAGCACCTCCAGCACAAACTGCGATGTGGGATCAGTTGAAAATGGATAGAGCATATTTTACTTCAAATGATGGTATTAATATTCTAGCAGGAACTGTCAAGATGTATGCTGGTGATCTAAAAAACACATCAAATACTGCACCAATCAATATAATATCAACAGGAGATATCGTTCTTGATGCCATGAACTATGATGTAAATATAGGGCAGTTAGATCTGATGTTACCAGCACAAAAGCTTCGAGATGAATTAGAAGCTAGCGGTAAAAAGGATTCTGTTACATTAAATACAATCAACCAGCTCAATGAAGAAATTGCATTTTATTTATCAAGATCATTAAATGGAACAAGAAGCCAAAGTAGCCATATTGATTCAGCACAAAATATTAATCTCCTATCTAAGAAGGGAATACTCATTAGAGCTTCAGAATTATATGCTAAAAATGAAGTAAATATTGAAGCACAAGGGTTATTAGCACGTGATCTTACAGGGGCTCAGGTAACTGATTATATCGATACTTCAATTTTAGTAGATGGGGTACATGATATTTATAAAAATGGTGAAGCTACTAATTCAAATTATGAAGAACGCTCTGATTTTCATAATTCAATTGTTAGTGGTGATAATGGAATCAATATTAAATCAACAGGTTCAGCCAAATTAAATTGGTATGATAATCCAAACCTATTATCTAATTACGCTACAGTCACTTCAGCAAAACCTATACAGTTAAAAAAGAAAATAAATGATGGAAAAAATAATATTGTTTTTAATGGTGCAGAATTAATTTCATCAGGAGGAGATGTCAATATTCAATCTAATGCAAATATTGTATTAGAATCAAGTCAAGATGAGATTTATAAGCATAGTACCAGTCAAGTTATAAAAAAAACTTGGTATGGAAAAAAGAAATCTACAGAAACCTATACACAGAGCCAATATGGGACAGCTGTTCCAACAACAATTTTAGCAAAAAATATTAGTCTGAAGGCAGCAGGAGATATAGATTTCTATTCAACGTTGATGAAAGCAAATAGTGGTAATGTTAATATTACTGGCGAAAATCTTTACTTTCTAACTTCAAATAACTATGCAAATTCAACTAGTACAACGAAAAAAAACTCATCAATTTTAGGAATTCCACTTAATAAATCTAAAACGACTAGTAGTCGTAGTCAAATTTCGCAACTTCCAGTAAAGCTGGTTGGAGATTATCTGTCAACTGAATCGAAAAATGATACAGTTTTTGTTGGGACAGAGTTTGATTATATGAAAAATGCGTCAGTTAAGGCAGGTGGGCAGATTGCTTTCCTTGGGGCATCAGAAACAATAAGTCAAGCTTTAAAAGTTGAAAAGAGCAATATTGTTTGGCAATCAATGCAAGATAAAGGCTCTATTACTGAAACTACTAAACTCCCAAATTTTAATGGACCTACTCCACCTACTTTTAAAGCATCAGGTGGCTTAATAGTACAGGTGCCTATTATATCTAATAAAAATAATGATATCCGTACAGAAGTGATTAACCTTGCTAATCAACCAGGTAATGAATATCTCAAAGGTTTTATTGCCAGAAAAGATGTAAATTGGGAGGCGGTTAAACTTGCACAAGAGAATTGGGATTATAAATCACAAGGTTTAACTGCTGCTGGTGCTGCAATTATTGTAATCATAGTTACAATTGCTACGATGGGTTCTGGTACTGCAGCTGCAGCTGGTGCTGCAGGTGGAACTGCTGCTAGTGGGACTACAGTAGGTCTTGGAGCAAGTATGATTGGAACTGCTGGTGTAACTACGACTGCAGCAGGAGCAATTGTTCCATCCACTTTAGGTGTTATGGCTAATGCAGCTGTTACAAGTTTAGCAACTCAAGCAAGTATTAGCTTAATTAATAATGGAGGTGATATTAGTAAGACACTTAAAGATTTAGGAAGTAAAGAAACTGTTAAAAATTTAGCAACTTCGGTTGTTACTGCAGGAGCCTTGAGTACAGTTGGTGGGCTTGATTGGATGGAAAAATTAAAAGATATGGGTACAGATCCGAATCTTTTAGTAAATCTAGCAGGGCGTACTGGTGGAGCTATAGTTGAAGCCGGTCTAAGTGCAGGAATTAGTAGTAGTATCAATGGTGGAAGTTTAACTGATAATTTACAAGGTATGTTACTTTCTAGTTTCGCAACAGCTTTACAGGGTAGTTTAGCGTCAAAAATTGGTAAAGAACTAGGAGGAATGAATAAAACAGATTTCGAAAATATTTTACATAAAATAGCCCATCTTGCTGCTGGGTGCGTAGCTGGTGCTATACAAAAACAATGTGAATCTGGCGCTATAGGTGCAGGAATTGGAGAGTTTGTCGCAGAGTTCATGCCAGCAGCCAGTGGCAATGGGGTGTATAGTGATGACGAAAAAGAAAGAGTTTTAGCAGTTGGTAAAATTTTGGCAGCTGGTGTAGCTGGTATTACTGGGTACGATGTAAACGTTGCATCAAGCTCTGCAAATACAGCACTGTTGAATAATGCATTATCAAAAACGGATGTAGATGTATTAATTCAAAACTTAAAAAGCTGCAAAAATGAATCACAATGTCGAAATGAGATTCAAAAAGCAGTTCTCAAGAGTAATTATAATCAAAGTAATGCAGTGAATTGTCTTTCGTTACTTTGCTCAGATAATCTTGAGCAGGCAATTAGTGGTTATAATCAGCTTAGAGGATTTATAGCAAGTGGTAAATTTTCAGGAGATAATTTAAAGCTATTAAATAATTTACTCACTCAAAGTACTAAAGATATTACTCTTCAAATTAATAGACTACCTCAAGTTCAAGCTTATCTAAATTCCCCTTGGACACCGCCAACAAAGCAACAGATGGCACAGATAGTTATTGATATTTTACCAATGACTAGTGATCCTTCTGCAATTTATTCAATTATTACAGGTAAAGCAGTTGTTACACAAGAAGAGGTAAGTCGTTTTTATGCAGCTGTTGGTTTAATTTTGCCAATTACAATTGGTCGTTCTTTAACAAAAGCCGAAATTGCTGGTTTGAAACAGTCTTCGCAAAATCCACTAAGCGGTACAGTAAGTATAAAAGATCTCCCAGCTCTACCAAAATCTGCACAAAACTTCGTTATTGAACAAACAGCACAATCTAAATATACAAAACTTTTATATGAAGCAAAGAATGATCCTAGTTTAGGAACTTGGAGTACTTCTAAAATAGGTCAAAAGGGTGAGGAAATAGCTGTACAGTTGGTTAAGGGGAGTGGTTTTACAGATGTTATAAGTATACAAAACGCTAGTGGTAATGGTATTGATATTATTGCAAAAAATCCTCAGGGAAATTATGTTTACTTAGAAGTAAAAACATCTGCTGTTGGAAAGATTGGTAGTTTATCTAAAAGGCAAGAAGATATGAACTTCTTTGTAAATGATATATTAACAAATGCCGCTGCTAAAACTGGGCGTTATGTAAACATAAGTGATACTGTAAATGCACAAGCTAAAATTATGTTAACAAAATATAAGCAACAAAAAGCTGTAGGTAATATCTATGGAGCAGCTATAGGGGTAGATTTAAAAAATGCTCAAATCTTAATCTCTTCTTGGACAAATAAATAA
>Cronobacter_turicensis_z3032_idrD
GTGTCTGAACCGTTAGCTGCCCGTTATCAGGACCCGCTCATCCACAGCTCTCTGCTCGCCGATGTCGTCAGCGGTGTGGTCGAAGGCGCCATTTGTCTTGCTGCGTTTACGGCGGGTGCGGCGATGATGACAACGGGGCTTGGCACGGTCGCAGGCGTTGTACTGATTGCCGCTGTTTTCGGCAGCGGCATCGCGGAAGAGGCAGGCGATCTGGTCGGCCAGGGTGTTGATGCCGTGCTGGATTTCTTCGGCTGGCGTGGGCCACCGGATGCCTTTATTACCAGTGGCTCGCACAACGTTCATATCATGGATCTCCCTGCCGCCCGTGCGGCGGGTACGGTCGATCATGATTATCTCAATACGCCTATCCCGGCAGAGAGCTTTGCCGATAAGGCGAAGGCGTTCGCGATAAACACGGCGGTTACCATTCTCGAAGTGGCGCAGTTTACGCTGCACCCGTTTGATAATCTGGCTGCGGGAGCCGACGCGATTGCGAGCAGCGGCTGGCAGGGCGTGAAAAATTTCGCGGGCAGCGTCTGGGATAACTTAACCCAGCCGGTCGTGGCGGGGGCCAGCCCTTTTGCTAAAGAAGCGCCGCTGGATACTGTCGAGTGCACCAAAGGCCATACGATCACCGGCGGTAACTTCCTGGCGGAAGGTTCGAAAAAAGTGCTGATCAACGGTCAGCCCGCGTGCCGTGATGGCGATCGCAGCACCTGCGAAGCAAAGATAAAAGTTAAAGAAAACACGCGTGTGCGCATCGGCGGCGAAAGCATTGTGGTGCGCGATATCCGCAGCGGTAAAAATTTCTGGGCGCGGCTTATCGGTAACGCCATCGGCAGCCTTGGACCGGGAATATTGCGCAATCTCAGTAAAGGGATGTTAAAGGCGATATTCAGCCGCCAGATCCTGAAAACCTTTTGCTGTCAGCTCGCCGCCGATCTCGGCATGGGATTAACCACGTTCGGGCTGATTCAGGCGGGGAAAGTGGGCAGCGAAGCGCGCCACACGCAGCACCCGGTGGATATCGCCAGCGGCGCGAAAATCCTCGCGGGCGGCGAAGACCGCGATTTCACGCTGGAAGACCGCATTCCGCTCATCTGGCAGCGTGTTTACAACAGCCGCAATCTGGCGACCGGTATGCTCGGTACCGGCTGGCTGCTGCCGTTTGAGACCCGCTTCTTCCGTCTTGAGAACAATCAGTTTATCTGGCGCGATATGTCGGGCCGCGATCTGGGCTTCGGCGAGCTGACCCCCGGCGACGTGGTGGATTATCTCGAAGACGGCGTCACGCTCTATTACACCGTCAGCGGCACGCTGATGCTCCAGATGACGAGCGGCGAATATCACGTCTACGAACCGGACCCGACGCGTCCGGGCGAGTGGCGGCTGTTCCGCATCTACGATCGTCACGAAAACTGCCAGTATTACAGCTGGGATGAACACGGCAGGCTGGCGCGCATTTCCGGCGACAACGAAGCGCTGGATGTCGAACTTGCCTACGAGAAATCGCATGGCCGTCTCGCCAGCGTGCACCAGGTATGCAACGGCGAGCGCCGCCTGCTGGTGACTTATGGCTATAACGAAAACGGCCAGTTGATCGAGGTCACCGACGCGGACGGCATCGTCACGCGCCGCTTCGGCTGGGATCGCGCCAGCGACATGATGGGCTGGCACAGCTACTCCACCAACCTCAGCGTGCATTATCAGTGGCAGCCCGCCGCCGATGCGCCCAACTGGCGCGTGTGCAGTTATCAGGTGCTGGACGACCAGGACAACGTGCTGGAGCGCTGGCGCATCGACGCCGACGAGGCGAAACGCTGCGCCACGGTAAGCTGCGACGCGGGTTTCTCGACGCGCCACTGCTGGGATTTCCTCTACCGCATCACGGAATACACCGACCGCCACGGCGGCGTGTGGCGCTACGAGTGGGCAGATTATGCCGAGCTGCTGAAAGCGGCCACCACGCCGGACGGCAGCCGCTGGGTGTATGGCTACGACGAGCACGGCAACCTGACGGAAGTGCGCGATCCGCTCGGCAACAGCACATTCACCACGTGGCACCCGGTGTTCGCGTTCCCGGTGAAGGAAGTGCTGCCGGACGGCGCCGCCTGGCAGTATGAATACAATGCGCGCGGCGATGTGGTGGCGCTCACCGACCCGAAAGGCGGCGTGACGCGCTTTGAGTGGAACGAGCAGGGCGACCTGGTTCAGCAGACCGACGCGCTGAACAACACGCACCGGTTCTGGTGGAACGAGCGCGGACAGCTGGTGCGCGACGAGGACTGCTCCGGCAACCAGAGCCAGCGGCTTTACGACGACGCGGGGCGGCCGCTCAGCGCCAGCGACGCCGAAGGCAACACCGACCGCTGGGCGCTGACCGCGGCGGGGCGTCTGCGCACCTGGCGACGGGCGGACGGCCGCGAGACGCACTACGAATATGACAACGCCGGGCTGCTCTGCGGGCAGGACGACGACGGGCTGCGCGAGCGCAAGGTAACGCGCAACGCGCGCGGGCAGGTGGTGAGCGCCGCCGACCCGGCAGGCCATCTGACGCGCCTGCGCTATGACCGTTTTGGCCGTCTGACGACGCTGGTGAACCCGAACCGCGAAAGCTGGCGGTTTGAGTATGACGCCGCGGGCCGCCTGACGGGCCAGCGCGACTACGCGGGCCGCCTGACGGAATACCGCCACGACGCGCTGGGCCAGGTGACGGAGGTGATCCGCCATCCGCTGCCGGGCAGCCCGGACGCGCCGCTGGTCACCGCGTTTGAATATGACGTGCTGGGCCGCCTGACTGCGCGGGAAACCGCAGAGCACCGCACCGAATACCGCCACGACACGCTGTCGCTGGAAATCCGCCGCGCCACGCGCGCCGAATGGCGGCAGGCGCTGCTGGAAGAGCGCGAACCGGCGTGGGACGCCGTGCTGGTCTTCACCCGCAATGCCGCGGGCGAACTGGTGAGTGAGGAGAACCACGGCGGGAAGTTTGAGTATGAGTACGACGCGCTGGGCAACCTCAGCAGTACAACGTTTCCGGATGGCCGGGAGCTCGCCACCCTGCGCTACGGCACCGGCCACCTGCTGGAGATGCAGCTGCGCCACGGCGGGGCCACGCACACGCTGGCGGCATACGGCCGCGACCGCCTGCACCGGGAAATATCCCGCAGCCAGGGTGTGCTCTCACAGGAGACCCGTTACGATTCCGCAGGCCGCGTCACCCAGCGCACGGTGCTGGATGCGCGACGCGAACTGGTGTTCGAGCGTCGCTACCGCTGGGACCGCACCGACCAGATAGTCCAGCAGATACACACTGACTCAACCCCTGCCACGCCGGGCGAGAAATACAGCCAGTACCTGTGGGGCTACGATGCCGCGGGCCAGGTCACAAAAGCTGTCGAACCGCAGAAAGAAGAGCGCTTCTTCTGGGACGCCGCGGGCAACCGCACTGAGGAGCACCGCAACCCGGTGTGGCACAACCTGCTGCTGCGCCTCGACGGACTGAAGCTGGATTACGACGGGTTCGGGCGACTCACGCGCCGTCAGGATAAAAGCGGCGTAGTGCAGCATTTTGCCTACGACGATGAGCAGCGGGTAAAAGAGATTCGGTTTGAGGGGAACACGAAGTTCCGCCGGGTGGAATACCGCTACGACCCGCTGGGGCGACGCACGCATAAAATCCTGTGGCGCTACGGAGAGAAAGACCCGGAAACCATCCGCTTCGACTGGCAGGGTTTACAGCTTGCAGGTGAACAGAGCGACCGGGAGCCTGACCACTACGTTCAGTACGTTTACACCGAAGGCAGCTACGAACCGCTGGCGCGGGTCGACAGTGTGTATGACGACTGTGAGATTTACTGGTACCACACCGAACTCAACGGCCTGCCGGAGCGGGTTACCGACGCGAACGGCCAGACCGTCTGGCGCGGACAGTTCAGCACCTGGGGCGAAACCGAACGCGAGCTCAGCGTGCCGCAGTGGCAGGTGCCGCAGAACTTACGCTTCCAGGGCCAGTACCTCGACCGTGAAAGCGGGCTGCATTACAACCTCTTCCGCTATTACGACCCGGTTGCCGGGCGTTACACCCAGATGGACCCGATTGGGCTTCTTGGTGGGATTAATACTTATGGATATGTGCCGGACCCATTAACTTGGGTAGATCCGCTTGGTCTGTGCTTCAGTAAGCGCTTGGGTGATTTTGGTGAATCCCATGTTAAATCTCAGCTCGAACGCTCAGGAAATTATGCAGATGTTTTTTCAGTGCAAAATAAATCTAATAACGGTATTGATTTAGTCGGCTTAAGACATGACGGGAAATATGACTTCTTTGAAGTAAAAACAAATACAACCGGATCTGTCGGCAAATTAAGTGATCGGCAGCTAAGTTCAAAAGATTTTATTGAAACTGTTCTTGGAACAGATGTCCGGAAAGGAGGCTATGACCTTAACGGGCACAGAGTCAATGATATGTTGGATAATATAGGTGATACTTATGTTGTCGATGTTTTCGTTGAAAGAGCCTCTAATGGGCGTTGGTTTGTTTCAAAGTATTTGATGAGTATTTGGGAGTAA
>Prevotella_jejuni_strain_CD3_33_chromosome_II_idrD
ATGGTGTGGGCGCGCGACGTGCTTGGGAACGTGACGGCCTACGCCTACGACGACGCCGACAACCTGATACACATTGAGGAAGCGGACGGCAACGTGCACAACCTCGTGTACGACGCCTCGGGTAACCTCGTCAAGGCGTCGGACCTGCTGCACGACGTGGAGTTCGACTACGGCCCGCTGGGCACGCTCACGGGCAGGCGGCAGTTCGGCCGCCACGTGCGTTTCGGCTACGACAGCGAACTGCAGTTGCGCGAGATTATCAACGAGGGCGGTGAATGCTATTCCTTCCGTCTTGACGGACTCGGTCGTGTCGTGGAGGAGACTGGCTTTGACGGCCTGCAGCGAAAGTACCTTCGTGATGGTGCTGGCAGGGTGATACGGGTTGTACGTCCCGAGGGAAGACAGACAGAATATGAGTATGATGGTGTGGGCAATATTCTGCAGGAAACACAGCATGATGGTCGAACAAGCCGCTTTGCTTATGACGGGGACGGACAGCTGCTGCGTGCTGAGAACAAGGAGATGAAAGTAGCCTTCAAGCATGACCTTGGGGGACGGATAGTGAAGGAGACGCAGGGCGGGCACTGCATCATGCGCAAGTATGACCGTACAGGACGCCATGTGCACACCGAGAGCAGTCTTGGTGCAAGTATTGACTATTCCCACGACAAGGATGGCAACCTCAGTGAGATGAACAGCGGAGGATGGTCAGCCAGATGGCAGCGTGACAGGGTAGGACTGGAGACGGAGCGTGAACTTACAGGCGGAGTGCATGTAAAGACACATCATGACAGCCTTGGACGTGAGACCTACAAGTCAGTGGGCGCACGCAATGTCGAGCAGTTCCGCCGCAGCTACACCTGGGGCATCGGCAACCGTCTTCATGCAACCGAGGACGGTCTTACGGGGCGCCGGGTACGCTATGACTATGATGAGTTCGACAATCTCCTTTCTGCAGAGTACAAGCAGGGGAATGACGTTGAGCGCATCTACCGTATTCCTGACCGTATCGGCAATCTCTTTGAGACACGTGAGAAGGATGACCGTAAGTACGGTGCGGGCAGCCGTCTGACGGAAGACCGTGACTACTTCTATCACTATGACTGCGAGGGCAACCTGGTGTTCAAGGAGTTCAGGACGCTGGACTGGGCCGGTGTGTCCGTTCCGCTTGGCGGACAGAAGGCGCACCTGGAGGAGGAGCTCGGCATAACCTTCCGTGCCTTTGGCGCAGGCTGGCGTTATGACTGGCAGAGTGATGGTATGCTGTCGAGGGTTGTTCGCCCCGACAGCAAGGAAGTGTCATTCGCATACGATGCGCTGGGCAGGCGGACCGAGAAGACTTATGAGGGCGTCGCTACTCATTTCGTGTGGAACGGCAATGTACCGCTGCACGAGTGGCAGGAAGTTGCAGCTGATGCCGGAAAGGCAGATGTCACCACCTGGCTTTTCGAGCAGAACACATTCATTCCTGCTGCCAAGCTCGCAGCCAACGGCGAGAGTTTCTCCATCGTGTCTGACTACCTCGGTACTCCCCTGCAGGCATTTGACAATAACGGCAACAAGGTTTGGGAGCAGGAGCTTGACATCTTCGGCAGAAAGAGAAGAAAGAACAACAATAATTCCTCCTTCATTCCGTTTAAGTATCAGGGGCAATATGAGGATGTTGAGACGGGGCTGTATTACAACCGCTTCCGCTATTATGAGCCGAATACAGGAACCTATATCAGCCAGGATCCGATAGGGTTGGTGGGCGGAAACCCGACACTATATGCGTATGTCGGAAGTCCTAACAACTGGTATGATATATTTGGCTTACGACCATTCGGACATGCTGTGGGAGATATTGGTGAGAAAGCTGTAATTAATGACCTTAAAAAGAACAATTATGAAATTATAGACGTAAAATACGGCTCTAACAATGGCATTGATGTGCTTGCCAAGAATCCAAGTACAGGAAAATATGATGCCTTTGAGGTAAAATCCTCTACGGTAGGAAAATTCAATCTTTCTGAAGCTCAGATGGATCCTGACAACTTTGTAAGGACACGATTGGAAAGAGCTGAATCACATGGTATGATTACCGAAGATGCTCGAATAGATATTATGTCTAACTTGGGAGAAAGAAAGGTTGCTTATGTGGATATAAAACGAAGAGATAATGGGGCTCTGTATGCTGAATCAATCCATTATGAGAGTTGGGATGCAGAGGCAAAAAGAATAGAAAAACTAAAAAAAGGAGGACATCATTAA
>Proteus_mirabilis_BB2000_idrD
ATGGGAAATGAATTTTTCGCAGCCAAGCAAGGCGATAAATTAATGCACTCGTCAATCTTCGCCGATATCGTGTGTGGTGTGGTTAAAGGGGCGGTTTATGCCGCGATAGGGACGGCCGCTGTGGCGTTGACTATTGGTACAGGTGGTATGGCAACGGCCGCCGGTGTTGCCATTTTTGCCGGTGCAGGGCTGGCGGCGGGCTTTACCATCGGTGGATTGATTGATGATGCCGCTGATTGGGTCGCGGATGGGATCGATAGCTTGTTCGGGCTAAGTAACCCTGATGGTGAAATCGATACTGGCTCGAAAAATGTACGTGTCAAAGGCAAGGGAGCCGCTCGCGCAGCGGGAAAATTACCGTCACCCGTACTATTAGCCTCAATTGCGGCAGATAATACCCCACCGAAAGAAGAAAAAGGCACCGAGATTGCCTTACGCGTATTGAAAAATACCGCCATGATGGCGACTGGGGCATTTTCTTTATTGGCTGGTAAGGTGCTAGGGAGCCAAACCAACAATGCGGCACCCATAGCGCTCGATCCCTCCGAGTTTCCCGAATACCCCTCACCGGAGCAAGGTTTTTTTGACAGTATTCTTTCCCCAGTAAAAGCTGCGAAACACCCGAATGCTGAACCTATGCCTTTGGATACCATCACGTGCGATAAATGGCATTGTGCTAGCGCCCCTTACTTTTTAGCCGAAGGGTCGAAAAAAGTCCAAATTAATAGCCAACCGGCGTGTCGTAACGGTGATAGAAGCACTTGTGAAGCCAAAATCAGTGATACGCAAGAAAAAGGGATTGTGCGTATTGGCGGTGGCAGTGTGGTGGTACGCGATATTATGAGTGGGCGTAACCCCTTTGCCGAAATGTTTGGTGAAATTCTCGGTGGCCTTGCTGTTGGAGCGGCGTCTACCCTGTTTCGCAGAGGGGGACTCAAGGCGATTAAACAGTGTTTGAATAGTGTAGGCTGTACTTTGGTGAGTGAAATCGCCAGTACCGCGGTGGGCAGTATGGTAGTCGCCTCGGTGACCCAAGCCTTTAATAGTGTGCGTCACCCCGTGCATGCGGCTACGGGGGCGAAAGTCCTAAATGGAGAGGATGATACCGATTTTATTATTGATGGTGTGTACCCGTTAGTCTGGTCGCGTATTTACCAAAGTCGTAATACGGGTGAAAATCGTTTAGGACGCGGTTGGTCGATGCCGTTTGATGTCTTTTTGACAATTGACGACACAGGTAAAGGGCTTGAGAATGAAAACATTTATTATCATGACATGTCGGGGAGGCGTTTAGCGTTAGGAAAAATCGCCCTTGGACAGAAAGTGTTCTATCAAGATGAAGGCTTTACCGTCTATCGCACGACAAACAATCTTTTCCTTGTTGAATCGGCGGAAGGTGATTATCAACTTTTCGAAGCCAATCCACATAAAATCAATACACTGCGCTTAATGAAAAGTGCTGATCGTCATAATAACGCTTTACACTACCGCTATGCTAATGACGGTGAATTGGTACAAATTCATGATGATGCGTATCTGACCGATATCCGGTTACATTATGATGAAATCACCCAACGCTTACAGTCGGTGACCCGCCACCAAGGACAAGAAGAAAAAACGCTGGTCACTTATACTTATGATGCACAACAGCGTTTAGTGCAAGTCACTAATGCGGATAAGCGAGTGACTCGTCGTTTTGGCTGGGATGATGAATCGGGTTTAATGGCCATGCACCAATATGCCACAGGGGTCAGTAGTCATTATCGTTGGCAACGTTTTGATGCCTTTACCATTGAAGACAATGAACCTGAGTGGCGAGTGGTTGAACACTGGCTTAAAGACGGTAAACGCTGTTTAGAGCATACCGAACTGACGTATGATTTAGCTCAACGAACCCTCACCACCGTGGAAACGGGGGGTGAAACCACCTTCCGTCGCTGGAATGAACAACAGCAAATTATCGAATACACCAATGCATTGAATGAAACGTGGTGGTTTGAATGGGATACCAGCCGCTTATTAACGAAAGCAATAGCCCCAGATGGCAGTGAATGGGGGTATACCTATGATGAGCGCGGTAATTTAACCCAATCGACCGATCCGGAACAACAATCGACTTGCTATGATTGGGACAAAGATTTTGCGTTTCCCACGGCACAAACTTTGCCAAATGGGGCGGCTTGGCATTGGGAGTACAATGAGCACGGCGATATTCGTCGTGTGATTGACCCGCTTGGCCATATCACGCGCTTGGCGTGGGATGACCAAGGCCTGTGTCTGGGGCAAGTGGATGCTAAGGGTAATGAAACCCACTATCGCTATAATGCCCGCGGTCAGTTAATCGAGCAACGGGACTGTTCGGGTTATCCCACCACCTTGACCTACGATGATTGGGGACAACTTCGCTCGTTGACCAATGCACAGAATGAAACCACGACTTACACCTTTAGTGAAGCGGGGTTGTTGTTAACAGAGCGCTTACCGGATGGGACAGAAAACCGTTATGACTATGATGCCACCGGGCAATTAGTGGGAATAACGGATGCCGGAGAGCGCCATATTCTGTTGCGCCGTAACCGTCGTGGACAAGTGATAGCCCGACGCGATCCGGCAGGGCATTGGTTGCATTTTCACTATGATACTTTCGGGCGCATGCAGGCACTGGAGAATGAACAGGGCGAGCAGTACCGCTTTGAGTATGATGCCTTGCATCGTTTAACCGATGAACATGATCTTATCGGACAACAAAAGCACTATCAGTACGATGTGATGGGGAATGTCACCCAGATAAAAACTACCCCGGGGCCTTGCGTTGATACGCCGATGCCCTTATCCCCGCTGGTGACCACCTTCGGTTACGATAAAGTGGGACGGTTGCTGTTTCGAGAAAACGCCGATTATCGCACGGAATACCTTTATCAACCTTTGAGTGTGACGTTACGCCGAGTCCCCATGGCGATATGGCATGAAGCCGAACGCACCGGCACCACCGCCCGCGTAGAGTATCAAGACGCCCTGACCTTTACCTACGATAAAGTGGGGCAGTTGGTTCGAGAAGCCAGTGCCCGCGATGATTATCAGCATCACTATGATGTGCTGGGTAATATCACTCGCACCGAGTTGCCCCATCAAAGGGCATTTGAATACCTGTATTACGGCTCTGGGCATTTACAACAAACGCAATGGCGAGATAATGCGCAACTGACGGTATTGGCGGAATATCAACGAGACCGTTTACACCGAGAAACGTTGCGCACCAGTGGCGCCTTAGACAATGAAACGGGCTATGACTGTCGGGGACGTATTACGCACCAAGTGGCGCGCCAAATGAATGCCTCACAGTTTGTGACCCCGGTGATTGACCGTCGTTATCGTTGGGATAAACGCAATCAACTTATCGAGCGCAGTGTCAGTTATGGTCAAACAGGCGAGGTTTTTACCGCCGGACATTGGTACTACCACAGTTATCAGTACGACCCGCTGGGGCAATTGACGGCGCATTTAGGGTCGGTGCAAACCGAACACTTTCTGTATGATGCGGCGGCCAATTTACTGACTCGTCCGCACAGCGAAGCACCGCATAACCAAGTACAAGGCAGTGATAAGTATGATTATCGCTACGACGGTTTTGGTCGCATGGTCTCCCGCTATGAAAAAGGCAGTAGCTCGGGACAACGTTATCACTATGACAGTGACCACCGCATTATCGCGGTGGATATCGACCAAGGGCCCTTAGGGTATCAGCGAGCGGAATACCGCTACGATATTTTAGGGCGCCGGATTGAAAAACGGTTATGGAAAGCCAGTGCGATTGCCAACACAGTCACCTATCACCAACATGAGCCGGATGAAGTGTACACTTTTGGCTGGGTAGGGATGCGACTGGTGTCTGAGCACAGCAGCGCAGCGCCCCATACCACGGTTTATCATGCATACAATGACCAAAGTTATACGCCTCTTGCTCGCATTGAATGCACGGATAACCCGCTTAATCCGCAACGGGCGATTTATTATACGCACAGCAGTTTAAGTGGCTTACCGGAAGCGTTGACCAACAGTGAGGGTGAAATAGTTTGGCAAGGGCAATACAGTGTTTGGGGACATTTACAGCGTCAAACCCGCCCCACAAGCACATTTAATCGTGAACAGAACCTGCGCTTTCAAGGGCAGTATTTCGACAAAGAGACGGGGTTACATTATAATACGTTCAGGTACTATGCCCCGGATTTAGGCCGTTTTACCCAGCAAGACCCAATAGGGTTGGCAGGGGGCATAAACTTATATGCTTATGCGCCAAATCCATTGACATGGGTGGATCCGTGGGGTTGGAGTTGCTTTAGTGCTCGGGTAGGTGCTTTTGGTGAGAAAAGAGTTATGAAATACTTATCTGGAGCGGGCTATAAAAAAGTTTTTTCTGTACAAAACAATTCTGGGCATGGTCTGGATATAGTTGCTTTAAGACCAGATGGAAAATTTGATATTTTTGAAGTTAAAAGTTCGACAATAGGACAATTTTCTTTATCTTCCCGCCAAGCTACAGGCGATGATTTTGCAAAAATAGTTCTTTTAAACGATGTGAAAAAAGGAGGTTATAATATTATCGATATAGATGGTAATGTTAAAGCAATTACAAGTAAACAAGCTAGATACATTTATAATAACATAGGAACAACCGAGTGGGTTCAGGTAAATGTTGGCCGAAATCCCAAATTTTATGATCAGAATATAACGTTTGAGGAATGGTAG
>Pseudomonas_fluorescens_F113_idrD
GTGCAGGACGGCAAGAATCGCCTGGTCACCGCCAGCCTCACCAGCCGCGACATCAAGAACAAAGCCGAGTACGACGCCAGCACCGTCAGCGTCGGCGGCGGCTATCGCGAAGTCGGCAAGGACCAGCAGGGCAACGCCACCAGCGGCGGCACGCAAACCCCTGGCACCGACCTGCCGAAGAACGACAACGACCTCAGCGCCACCATGCCGATCGCCATCAGTGCCAGCGACGATGCCAGCAGCGTCACCCGCAGCGGCATCAGTGGCGGCACCATCGTCATCAGCAACGACGCCGAGCAACAGAAGCGCACCGGCAAGAACGGCGAGCAAACTATCGCCAGCCTCAACCGCGATGTCTCCAGCGACCGCGACACCACCAACACGCTCAAGCCGATCTTCGACGAAGACGAAATCCGTGCCGGCTTCGAGATCGTCGGGGCGTTCGCCAACGAAGCCAGCACCTTGCTCGCCAACAAGGCCAAAGAAGTCGACCTCAAGCGCAAACAGGCCGAGGCGGCTCAGACAGCGGCCCTGGACCCCAACGCCAAACTCACCGACACCGAACGCCTGGCGCTGCTCGGCAGCGCTCAGCGCCTGAACGCCGAAGCCAGCGGCATCGCCGACAAGTGGGGCGCGGGCGGTACCTACCGGCAGATCACCACCGCCCTGGTGGGCGCGGCCAGCGGCAACATCAGCGGCGGTAACAGTGCCTTCGTTCAAGGGCTGGTGGTCAATTACGTGCAGCAGCAAGGTGCCGGCTACATCGGCGACCTGGTGGCGGACGGCAAACTCACCGAAGGCTCGCCGCTGCACGCCGCCCTGCACGGCATCGTCGCCTGTGCCGGCGCCGCCGCCAGCAGCCAGAGCTGCGGCAGCGGTGCTGCAGGCGCGGCGGCGTCGAGCCTGCTCACCGGCCTGTTCTCCGAAGCCAGCCCCGATGAAAGCGAATCCCAACGTGAGGCCAAGCGCAACCTGATCGTCAGCATCGTCACCGGGCTGGCCGCCACCAGTTCCTCGCTCGACCCGGCCACCGCCACCAGCGCGGCCGGGGCAGCGGTGGACAACAACTGGCTGGCGACTCAGCAGTTGGTTCAGGCGGAGAAGGAATACGACGCAGCTGACGGCTTAGGGGCCAAGGCGCAAGTGATCGCCAAGTGGGCGTACATCAATCAGAAACAAGACGTCCTGACCCGCTTCGGCGTGGGCAAAGGCTTGGTGCAGGCGGGGTGGAGCGACGTCGAAGGCTTGGCGCAATTCCTCCTTCACCCGGTTGATGGAATCAACGGCCTCAAGGCCCTGATCAACGACCCCGAGGCTCGCAAGCAGTTTGGCGATGGCGTGGTCAACGAACTCAACGCCAAACTTGACCGCGTTAAAGACGCCCTGGAAAACGGTGGTGATGATCGCGCCGTGCAACTGGGCCAAGACATTGGCGAACTCGTCTGGCAGGTCGGCAGCATCGCCACCGGTGTTGGCGGGGCGGCCAAGGGTGGCGTGGCGTTGGCGAAAGCCGGGATCAAGGTGGGGGCGGAAGGGCTTGAAAAAATGGCCGATATGGCCAGGCTTGAAAAAATAGCGAAAGTCGAAGCGCCAGCTACCAAGCCATTGCCTGACTGGAGCGGTGATGGACCCTCCGTCGGGAAGGGGCCAGATAAAACAGTCCCTCCGAGCACGACAACTGATCTGTTTGAAGCGTCTAGAGGTAAGGATCTTTCGACCCTGACAAACCAGCAAATTGGCGACTTGGGTGAGGCGATATCCAAAACCTTCTTGCGCGATAATAACCATACTGACATCTTTGCTATCCAGAACAGTAGCGGAAATGGCATCGATATTGTCAGTCGCACACCTGATGGTCGTCTCGCCTTCACTGAGGTGAAAACATCGCGAACAGGGAGTATTGGAGGGCTTTCCAGTCGTCAAGAAAATATGACGACGTTTGTCGAGGATATTCTGACTCAGGCAGCTAGTCGCCAGGGGCGCTACCGGAATATTGACGTCGCTGATCAACAGCGGGCACGTCAGATGCTCAGGGAGTTTAGGCAAACACCTGAAAGTGTGAGCGGAAATTTGATCGGTGTTGATCTAAAGGGAGAAGTATTGAGAGTCTCTCCTTGGGCGCGCAATTGA
>Rothia_C6B_idrD
atgaccgacatccacggcaatctcttatggtacagcgaatacaccgcctggggtcgtctgaaaaaggacgggcgggtttacaagaacgcgcaccagccgttcagattgcagaaccagtattacgatgaagagaccgggctgcattataacttgttgcgttattacgagcctgaggctgggcggtttgtgaatcaggatccgattgggttgttgggtggggagcatctttatcaatttgcagataacgctttggtttggttcgacccgttgggattgaaaaaaacctacgtgcagcgcttgggtactgccggcgagcgcagggtaatgaaatatctggaaggtacgggatacaaaaaagtcttctctattcaaaatgcctccggaaacggcttggatattgtcgcattgagaccggatgggaaatatgacatatttgaagttaaaagttctaaacgtggaaaatttaaactaagcgaaagacagcagaaaggcggtaaatgttttgctgagcaagtgttgacggaagatgtaacggataagaaaaaaggcggatattttatgaaaggcttagacggcaaaaagacgcctcttaataagaagaaggcgcaagagatatttaataatatagataaaaccgaaactgtctttgttgatatgaatcataaatttcaagcaacccgcatgacatttagcccatggtaa
>Xanthomonas_citri_pv_malvacearum_strain_XcmN1003_idrD
ATGGCCAGCACCACCGAGATCGCAGCACTGCTGTCTGCCACCGATGCACAGGGCCGGGTCGATCCGATGGCCTACGAAGCGCAACGGTTGATCCTGCAGCACCAAGGCCTGGGGAGCGTGGATGCTGCTGCGCTGGTGCATGCTCTGCCGGCCTCGCCGGGATTCGCCACCTTCGACCGTAACGCCTTCTTTTCCGCCATCGACCAGCGCCTGGAGACGCCGCAGGAACGGCAGCGCTTTGCTGATGCGCTGGACCAGGCCAACCTCAGCGATAGCTTGCTCGAGCGGCTGGGCGAGCGGGCCTTTGAGGCCGGTGGGCAGGCCTATGACGCTACGCGCAACGCGGCGAGCCGGGCCGGCGCCCAAGTCAGCGATGGCCTGGCCACTGCGCAGCGCACCGCCCGCCACCCGGCCGCCGATCCCGGTGCCTCGCCCGCGTTGCGCGCTGCAGCGAATGTCGGCAGCAACGTGATCGGTGAGGCTCAGCAAGGCTACGGTTTCGTCACCGGCGGTGCCACGCACGCCCTGTCCACGCTCAGCGATACCGTCGATCTGGCGCGCATGGGTGTGCGCTTGGCGTCCGATCCCAACTACCGAGACGTGGTGGTCGGCATGGTCCGCATCTATGCCGCCGAGGTCGCCGCCGACCCGCACAAGCCGATCGACCAGGTCCGCAGCGCCGTCACGCAGGCGTGGCAGCATTGGGAACAGGGGTTGGCACAGGCGCAACGCGATGGGCGCGAGCGCGAATACATCGGCAGCGCGACCGGTGCGGCCGGAGTGGAGATCCTGGCCACCCTGGTGCCGGCAAGCAAAGTGAGCAAGCTCGGCAAGGCCGCGCAGATCATGGAGGACATCGCCCCCCGGGCCGCCGCCGAGGCCAGCGAAGGCCTGGCCGATGCCGCGCGTGCCGAACGCGCTGGTGCGCGCGCCCACGACGGCACTGAGTATGCGCCCGAGCGCCATGCGGGCTTGCCCGGCCGCGCTCGCGCCTACGCCGAGGGGCTGGAGGAAATCGGCCAGGCCGCCCGGCGTGGTGGTGAGGCCGGCGAAGGGGCGCAGCAGATGCTGCGCGGGCTGGTCGGCCAGGCCCGCCAGGACGGCCACCTGGACGATCTGGTCCGCGCCGCGCGCGCCACCGACAACGTCGAAGGCCTGCTGCGCAGCGGCCAACTGGAACCGCACGAACTCACCGCCATCCTCAAGCGCACGCCGGGTGTCTTTGACGGCAGGATCGATTTCCCGACGGCCCTAGACCACAGTACGCAGGGCGTGGACCTGACCCGGCTCACCACCCGCCAGCTCGGCGACATCGGCGAAGCCATCCAGACCCATGACCTGGTAAAGCAGGGATATAGCGACATCGTCGCGATCAAGAACCGCTCCGGTCACGGCATCGACCTGGTCGGCCGCAACCCCGACGGTGATCTGGAGTTCTTCGAGATTAAGACCTCGGCCAAGGGATTGGCGCCGGCGCAACATGGGGACCCGGAACAATTCGTAGCCAAGCGGCTTGAGCGGGCGATTGACGCCCAGGGTCACTGGGATCCGAAGAACACGATACCTGGGCTGAAGTCGATCGCTCAGGATATCAGCAGGGAAATCGGCCCGGAGGCCGAGAACATCAACGCCACGTGGGTGCGGCTCAATCTGAGCCGGAGCCCGGACGCGCCGCACTTGCAGATCGAACGCAAGATAGATCCATGGGGCAAGCCCGCAGCGCCGAAACAGACGATGCTGACTCCTGGCGATGCCGACCACCCAGACCACGCCGCATTTGACACGATACTGCGCACGGTGCAGGGCGACGGCCGTTGGAACGTTGCGGAAAGCCACAATATTGCCGCGAGCCTGCTGAAGGAATACAAGGCCGATCCGCTGTGTAAGACCCTGGACTCGGTGCGCATCGGCGGCAGCGCCGATACGCCATGCATCTTCGCGGTCAGTTCGCCACACGGCGACAGAGGCCCGGATTTCCATGTCCACGTCCAAGCCGAGCAAGCTGCGCAGCGCCCGGCGGCCGAGAGCTTCGCTCAGGTCCAACAGATCAACCAGCAACAAGCGCTGACCCAGGAAACAGAGCAACAGAACCTGGCTCACCGCGGCCCGAGGATGAGTTGA
We put these sequences in a file called idrD.fna.
Identifying metagenomes likely to contain populations with idrD
Since there are too many metagenomes to practically download and query all, we screened for metagenomes likely to contain our sequences (or similar sequences), using SearchSRA. Briefly, SearchSRA employs the PARTIE algorithm (Torres et al 2017) on subsamples (100,000 reads per metagenome) from all metagenomes on SRA (which mirrors ENA and other international short read databases) and queries them with bowtie2. We uploaded the FASTA containing all 7 idrD nucleotide sequences to SearchSRA and searched against all metagenomes.
This resulted in a zipped folder with all the alignments. It was called
results.zip; we renamed it to searchsra_results_20190113.zip and
then unzipped it with the command unzip searchsra_results_20190113.zip
and then entered the resultant directory with
cd searchsra_results_20190113.
Now, we wanted to find which metagenomes (that is, the *.bam files produced from PARTIE) had non-zero, or practically zero coverage. Since PARTIE works by only mapping a [small] subset of reads from each metagenome, the actual mapped counts are not informative, other than whether any reads mapped (meaning that metagenome sampled a population with idrD) or didn’t map. For this first pass filtering of metagenomes, we worked off of file size, as metagenomes with zero mapping were 4KB, so we kept anything above 4KB. We did that with this one-liner:
for file in $(ls | grep "^[0-9]*$"); do
du -h $file/* | grep -v '4.0K'; done | awk '{print $2}' | sed 's/.bam//; s/^[0-9]*\///'
> sra_run_accessions_gtt4KB.txt
This resulted in 3801 metagenome accessions. We downloaded the available metadata for each of these metagenomes by looping through each entry like so:
for entry in $(cat sra_run_accessions_gtt4KB.txt); do
esearch -db sra -query $entry | efetch -format runinfo >> sra_metadata_accessions_gtt4KB.txt
done
This comma-separated file has the needed data but also junk whitespace (empty lines) so we cleaned that and converted to a tab-separated file by running
grep -v '^$' sra_metadata_accessions_gtt4KB.txt | tr ',' '\t' > temp
mv temp sra_metadata_accessions_gtt4KB.txt
We took this list and manually curated to remove genome assemblies, metagenomes with too-specific library preparation methods (e.g. target capture), etc. to ultimately produce a list of 1,189 metagenomes, including both single- and paired-end sequence libraries. This table can be found at this Drive link. In this table, the sample site for each metagenome is listed in the column “ScientificName”, with varying information content depending on the uploader.
Preparing the metagenomes
Downloading and sorting
We copied the curated list of 1,189 accessions, just the accessions with
one accession per line, to a text file called
curated_sra_list_20190114.txt and uploaded it to the cluster. The
first few lines of the file look something like this:
SRR1182872
SRR948162
SRR1291146
SRR1291147
SRR948162
SRR1291146
SRR1291147
SRR948163
SRR1291148
SRR1291149
Note: Everything from here until the end of Retrieving the coverage and variability data, occured on Harvard’s Odyssey computational cluster, due to the large filesizes and computational resources. Odyssey uses the Slurm workload scheduler; batch scripts copied below retain all header information for maximum reproducibility.
We looped through this list of metagenomes to download each one in parallel batches, using the following script submitted to Odyssey:
#!/bin/bash
#SBATCH -N 1
#SBATCH -n 1
#SBATCH --contiguous
#SBATCH --mem=1G
#SBATCH -t 0-01:00:00
#SBATCH -p shared
#SBATCH --job-name="downloadMaster"
#SBATCH -o odyssey_downloadMaster.out
#SBATCH -e odyssey_downloadMaster.err
#SBATCH --mail-type=NONE
# chunk the accessions into batches
cat curated_sra_list_20190114.txt | split -l 20 -d - sraBatch_
# get the batches
batches=$(ls sraBatch_*)
# write and submit the script that does the downloading, for each batch
for batch in $batches; do
echo "#!/bin/bash
#SBATCH -N 1
#SBATCH -n 1
#SBATCH --contiguous
#SBATCH --mem=1500M
#SBATCH -t 0-06:00:00
#SBATCH -p shared
#SBATCH --job-name='$batch-tar'
#SBATCH -o odyssey_unstuff-$batch.out
#SBATCH -e odyssey_unstuff-$batch.err
#SBATCH --mail-type=NONE
module load sratoolkit
for sra in \$(cat $batch); do
# fastq-dump does the downloading, --split-3 dumps 'junk' or unpaired reads a 3rd file
fastq-dump --split-3 $sra
done
" > slurm-$batch.sh && sbatch slurm-$batch.sh
sleep 30
done
Some details about this script:
- The lines starting with #SBATCH are instructions to the cluster we used (which uses Slurm)
- This is an ad-hoc parallelization, where the first part does not
actually download but splits the list up into batches of 20
accessions and then generates and submites a script for each batch
to actually download the metagenomes.
- Note: This could be written more efficiently as a job array, but our cluster was unreliable at this time so retaining individual batch’s scripts was useful for resubmitting stalled jobs.
This resulted in 1,188 metagenomes (a single metagenome failed to download; attempts to download individually failed repeatedly). The next step was to separate paired-end sequences from single-end sequences as the mapping program handles them differently. We accomplished this with a small script:
#!/bin/bash
mkdir single_end
mkdir paired_end
# move paired ends, clean off 3rd file of errors
for r1 in $(ls *_1.fastq); do
# extract the ID
prefix=$(echo "$r1" | sed 's/_1.fastq//')
# move the PE files to paired_end
mv $r1 paired_end
mv ${prefix}_2.fastq paired_end
# delete any 3rd errors
rm ${prefix}.fastq
# make list of which samples were pe and where to find them
echo "paired_end/$prefix" >> sra_paired_end.txt
done
# move single fastqs leftover
# make list of which samples were se and where to find them
ls *fastq | sed 's,.fastq,,; s,^,single_end/,' >> sra_single_end.txt
mv *fastq single_end
Mapping
Now that everything was set up, we mapped the metagenomic short reads to
the reference idrD sequences. We chose to use bowtie2, an accurate and
fast gap-sensitive read aligner (Langmead & Salzberg 2012). The first
step is to turn the FASTA file of idrD nucleotide sequences into a
bowtie2-compatible reference database, carried out with the command
bowtie2-build idrD.fna idrD. Now, we can map each metagenome against
the idrD reference database using the following scripts:
#!/bin/bash
#SBATCH -N 1
#SBATCH -n 16
#SBATCH --contiguous
#SBATCH --mem=32G
#SBATCH -t 1-12:00:00
#SBATCH -p shared
#SBATCH --array=0-32%33
#SBATCH --job-name="bt-pe"
#SBATCH -o odyssey_map_pe_%a.out
#SBATCH -e odyssey_map_pe_%a.err
#SBATCH --mail-type=END
# split -l 20 -d -a 4 sra_paired_end.txt pe_batch-
# mkdir pe_bt_mapped
style="pe" # pe = paired end, se = single end
module load samtools bowtie2 zlib xz
taskNum=$(printf %04d $SLURM_ARRAY_TASK_ID)
# get list of sample-runs, chop into per sample batches
batch=${style}_batch-$taskNum
# iterate for each participant
for samp in $(cat $batch); do
sampOut=$(echo "$samp" | sed "s,^[a-z_]*,${style}_bt_mapped,")
R1=${samp}_1.fastq
R2=${samp}_2.fastq
# map sample set
bowtie2 -x idrD -1 $R1 -2 $R2 --no-unal -S $sampOut.sam --threads 16
samtools view -S -b $sampOut.sam > $sampOut-RAW.bam
samtools sort $sampOut-RAW.bam > $sampOut.bam
samtools index $sampOut.bam
# clean as you go
rm $sampOut.sam $sampOut-RAW.bam
done
The above script was for the paired-end metagenomes, and the script below was for the single-end metagenomes:
#!/bin/bash
#SBATCH -N 1
#SBATCH -n 16
#SBATCH --contiguous
#SBATCH --mem=32G
#SBATCH -t 1-00:00:00
#SBATCH -p shared
#SBATCH --array=0-54%20
#SBATCH --job-name="bt-se"
#SBATCH -o odyssey_map_se_%a.out
#SBATCH -e odyssey_map_se_%a.err
#SBATCH --mail-type=END
# split -l 20 -d -a 4 sra_single_end.txt se_batch-
# mkdir se_bt_mapped
style="se" # pe = paired end, se = single end
module load samtools bowtie2 zlib xz
taskNum=$(printf %04d $SLURM_ARRAY_TASK_ID)
# get specific batch
batch=${style}_batch-$taskNum
# iterate for each sample
for samp in $(cat $batch); do
sampOut=$(echo "$samp" | sed "s,^[a-z_]*,${style}_bt_mapped,")
# map sample set
bowtie2 -x idrD -U ${samp}.fastq --no-unal -S $sampOut.sam --threads 16
samtools view -S -b $sampOut.sam > $sampOut-RAW.bam
samtools sort $sampOut-RAW.bam > $sampOut.bam
samtools index $sampOut.bam
# clean as you go
rm $sampOut.sam $sampOut-RAW.bam
done
IMPORTANT: Before running these scripts, as they are job arrays, we
ran the first commented-out lines
split -l 20 -d -a 4 sra_single_end.txt se_batch- and
mkdir se_bt_mapped for the single-end metagenomes, and the
corresponding lines for the paired-end metagenomes. These commands first
make the lists of files (with 20 accessions per file) that will be
processed by each instance of the job array and secondly make the
directory into which the mapping results will be placed.
Importing the results into Anvi’o
We managed the data with anvi’o, a platform for analysis and
visualization of ’omics data (Eren et al.,
2015). The first step is to make a
so-called contigs database to hold the sequence information, which we
accomplished with the command
anvi-gen-contigs-database -f idrD.fna -o idrD-CONTIGS.db --skip-gene-calling
Now we can profile the contigs database with the bowtie2 results, which will make a so-called profile database that stores and summarizes the bowtie2 mapping results in a readily accessible manner. For the paired-end metagenomes, we used this script:
#!/bin/bash
#SBATCH -N 1
#SBATCH -n 4
#SBATCH --contiguous
#SBATCH --mem=2G
#SBATCH -t 0-01:00:00
#SBATCH -p shared
#SBATCH --array=0-32%33
#SBATCH --job-name="prof-pe"
#SBATCH -o odyssey_prof_pe_%a.out
#SBATCH -e odyssey_prof_pe_%a.err
#SBATCH --mail-type=END
# split -l 20 -d -a 4 sra_paired_end.txt pe_batch-
# mkdir pe_profs
style="pe" # pe = paired end, se = single end
# activate anvio
source ~/virtual-envs/anvio-dev-venv/bin/activate
module load samtools bowtie2 zlib xz
taskNum=$(printf %04d $SLURM_ARRAY_TASK_ID)
# get list of sample-runs, chop into per sample batches
batch=${style}_batch-$taskNum
# iterate for each participant
for samp in $(cat $batch); do
bam=$(echo "$samp" | sed "s,^[a-z_]*,${style}_bt_mapped,")
profOut=$(echo "$samp" | sed "s,^[a-z_]*,${style}_profs,")
# make anvio profiles, -M 0 means don't skip profiling contigs less than 2.5kb
anvi-profile -i $bam.bam -c idrD-CONTIGS.db -o $profOut -M 0 -T 4 --write-buffer-size 700
done
Note that like before, the split and mkdir commands commented out in
the script were run at the command line prior to submitting this array;
likewise for the single-end metagenomes but substituting single and
se for paired and pe, respectively.
For the single-end metagenomes, we used the same script but changed the
line style="pe" to style="se"
This resulted in two directories, pe_profs and se_profs, each with
hundreds of profiles, one per metagenome, that contained all the
coverage and variability information we are interested in. To facilitate
future steps, we used the anvi-merge command to merge the indiviual
profile database into a single merged profile database with the
following script:
#!/bin/bash
#SBATCH -N 1
#SBATCH -n 1
#SBATCH --contiguous
#SBATCH --mem=12G
#SBATCH -t 0-04:00:00
#SBATCH -p shared
#SBATCH --job-name="merge"
#SBATCH -o odyssey_merge.out
#SBATCH -e odyssey_merge.err
#SBATCH --mail-type=END
# activate anvio
source ~/virtual-envs/anvio-dev-venv/bin/activate
se=$(cat sra_single_end.txt | sed 's/^[a-z_]*/se_profs/')
pe=$(cat sra_paired_end.txt | sed 's/^[a-z_]*/pe_profs/')
# make anvio profiles, -M 0 means don't skip profiling contigs less than 2.5kb
anvi-merge $se $pe -c idrD-CONTIGS.db -o idrD-MERGED -M 0 --skip-concoct-binning
Retrieving the coverage and variability data
Now that all the data was together, we extracted the coverages and
variability information per site per gene for each metagenome. To do
this, we made a dummy collection to keep anvi’o happy with the command
anvi-script-add-default-collection -p idrD-MERGED/PROFILE.db. Then, we
exported the coverages with
anvi-get-split-coverages -c idrD-CONTIGS.db -p idrD-MERGED/PROFILE.db -C DEFAULT -b EVERYTHING -o idrD_covs.tsv
and the variability information with
anvi-gen-variability-profile -c idrD-CONTIGS.db -p idrD-MERGED/PROFILE.db -C DEFAULT -b EVERYTHING --include-split-names -o idrD_var.tsv
Here are first 10 rows of the coverage data:
nt_position split_name sample_name coverage
0 Acinetobacter_baumannii_strain_XH858_idrD_split_00001 ERR1121387 0
1 Acinetobacter_baumannii_strain_XH858_idrD_split_00001 ERR1121387 0
2 Acinetobacter_baumannii_strain_XH858_idrD_split_00001 ERR1121387 0
3 Acinetobacter_baumannii_strain_XH858_idrD_split_00001 ERR1121387 0
4 Acinetobacter_baumannii_strain_XH858_idrD_split_00001 ERR1121387 0
5 Acinetobacter_baumannii_strain_XH858_idrD_split_00001 ERR1121387 0
6 Acinetobacter_baumannii_strain_XH858_idrD_split_00001 ERR1121387 0
7 Acinetobacter_baumannii_strain_XH858_idrD_split_00001 ERR1121387 0
8 Acinetobacter_baumannii_strain_XH858_idrD_split_00001 ERR1121387 0
And here are the first 10 rows of the variability data:
entry_id unique_pos_identifier pos pos_in_contig split_name sample_id corresponding_gene_call in_partial_gene_call in_complete_gene_call base_pos_in_codon codon_order_in_gene codon_number gene_length coverage cov_outlier_in_split cov_outlier_in_contig A C G N T reference consensus competing_nts departure_from_reference departure_from_consensus n2n1ratio entropy
0 0 155 155 Prevotella_jejuni_strain_CD3_33_chromosome_II_idrD_split_00001 DRR046092 -1 0 0 0 -1 0 -1 13 0 0 0 9 4 0 0 C C CG 0.3076923076923077 0.3076923076923077 0.4444444444444444 0.6172417697303416
1 0 155 155 Prevotella_jejuni_strain_CD3_33_chromosome_II_idrD_split_00001 DRR046097 -1 0 0 0 -1 0 -1 14 0 0 0 10 4 0 0 C C CG 0.2857142857142857 0.2857142857142857 0.4 0.5982695885852573
2 0 155 155 Prevotella_jejuni_strain_CD3_33_chromosome_II_idrD_split_00001 SRR1284495 -1 0 0 0 -1 0 -1 35 0 0 0 31 0 0 4 C C CT 0.11428571428571428 0.11428571428571428 0.12903225806451613 0.355382896246011
3 0 155 155 Prevotella_jejuni_strain_CD3_33_chromosome_II_idrD_split_00001 SRR3067121 -1 0 0 0 -1 0 -1 18 0 0 0 14 2 0 2 C C CG 0.2222222222222222 0.2222222222222222 0.14285714285714285 0.6837389058487535
4 0 155 155 Prevotella_jejuni_strain_CD3_33_chromosome_II_idrD_split_00001 SRR3067123 -1 0 0 0 -1 0 -1 27 0 0 1 21 2 0 3 C C CT 0.2222222222222222 0.2222222222222222 0.14285714285714285 0.7544627023259549
5 0 155 155 Prevotella_jejuni_strain_CD3_33_chromosome_II_idrD_split_00001 SRR3067131 -1 0 0 0 -1 0 -1 36 1 1 1 32 2 0 1 C C CG 0.1111111111111111 0.1111111111111111 0.0625 0.4643566259363562
6 0 155 155 Prevotella_jejuni_strain_CD3_33_chromosome_II_idrD_split_00001 SRR3067135 -1 0 0 0 -1 0 -1 54 0 0 0 49 4 0 1 C C CG 0.09259259259259259 0.09259259259259259 0.08163265306122448 0.3548290085661212
7 0 155 155 Prevotella_jejuni_strain_CD3_33_chromosome_II_idrD_split_00001 SRR3067140 -1 0 0 0 -1 0 -1 13 0 0 1 9 1 0 2 C C CT 0.3076923076923077 0.3076923076923077 0.2222222222222222 0.937155853065701
8 0 155 155 Prevotella_jejuni_strain_CD3_33_chromosome_II_idrD_split_00001 SRR3067141 -1 0 0 0 -1 0 -1 61 1 1 0 57 1 0 3 C C CT 0.06557377049180328 0.06557377049180328 0.05263157894736842 0.27891059970489007
The coverage and varibility files were copied down from the cluster and then plotted with R, described in the next section.
We also ran these analyses for Prevotella spp., described later on in this document, but the coverage data here and variability data here are here as the code below will process both at the same time.
Inspecting metagenome coverage
Preliminary data processing
First, we read in the data like this:
library(ggplot2)
library(reshape2)
library(plyr)
library(dplyr)
library(tidyr)
library(entropy)
reads_path <- "~/idrD_analysis/idrD_covs.tsv"
var_path <- "~/idrD_analysis/idrD_var.tsv"
metadata_path <- "~/idrD_analysis/sra_metadata_accessions_gtt4KB_mgenomes_abbrev.txt"
prevo_cov_path <- "~/idrD_analysis/prevo_idrD-covs.tsv" # this is for later
prevo_var_path <- "~/idrD_analysis/prevo_idrD-var.tsv" # this is for later
covs <- read.table(reads_path, header = T, sep = "\t")
vari <- read.table(var_path, header = T, sep = "\t")
# check out the first few lines
head(covs,3)
head(vari,3)
## nt_position split_name sample_name
## 1 0 Acinetobacter_baumannii_strain_XH858_idrD_split_00001 ERR1121387
## 2 1 Acinetobacter_baumannii_strain_XH858_idrD_split_00001 ERR1121387
## 3 2 Acinetobacter_baumannii_strain_XH858_idrD_split_00001 ERR1121387
## coverage
## 1 0
## 2 0
## 3 0
## entry_id unique_pos_identifier pos pos_in_contig
## 1 0 0 155 155
## 2 1 0 155 155
## 3 2 0 155 155
## split_name sample_id
## 1 Prevotella_jejuni_strain_CD3_33_chromosome_II_idrD_split_00001 DRR046092
## 2 Prevotella_jejuni_strain_CD3_33_chromosome_II_idrD_split_00001 DRR046097
## 3 Prevotella_jejuni_strain_CD3_33_chromosome_II_idrD_split_00001 SRR1284495
## corresponding_gene_call in_partial_gene_call in_complete_gene_call
## 1 -1 0 0
## 2 -1 0 0
## 3 -1 0 0
## base_pos_in_codon codon_order_in_gene codon_number gene_length coverage
## 1 0 -1 0 -1 13
## 2 0 -1 0 -1 14
## 3 0 -1 0 -1 35
## cov_outlier_in_split cov_outlier_in_contig A C G N T reference consensus
## 1 0 0 0 9 4 0 0 C C
## 2 0 0 0 10 4 0 0 C C
## 3 0 0 0 31 0 0 4 C C
## competing_nts departure_from_reference departure_from_consensus n2n1ratio
## 1 CG 0.3076923 0.3076923 0.4444444
## 2 CG 0.2857143 0.2857143 0.4000000
## 3 CT 0.1142857 0.1142857 0.1290323
## entropy
## 1 0.6172418
## 2 0.5982696
## 3 0.3553829
The file at metadata_path is the metadata downloaded earlier from NCBI
and is the file we used earlier to get the curated metagenomes list, but
we trimmed off some columns here that we did not use.
Next, we read and processed the metadata to identify which metagenome
came from which category. The metadata called the HMP datasets only as
“human metagenome”, not separating them by body site, but that
information was stored in the “AnalyteType” column. So, we updated the
“ScientificName” column, which held the desired information for all the
other metagenomes, to reflect whether the metagenome was HMP oral or
otherwise. We also manually looked through the “human metagenome”
samples to identify other oral metagenomes that weren’t from the HMP. We
added a column called “category” to the covs and vari dataframes to
hold this information.
# drop unique "Run" id's
metadata <- distinct(read.table(metadata_path, header = T, sep = "\t"), Run, .keep_all=T)
rownames(metadata) <- metadata$Run
# convert to character to be safe
metadata$ScientificName <- as.character(metadata$ScientificName)
# rename the HMP gut samples' ScientificName
metadata$ScientificName[as.character(metadata$Analyte_Type) ==
"G_DNA_STOOL"] <- "HMP gut metagenome"
# likewise for HMP oral
metadata$ScientificName[metadata$Analyte_Type %in%
c("G_DNA_Tongue dorsum","G_DNA_Throat")] <- "HMP oral metagenome"
metadata$ScientificName[metadata$Run %in%
c("SRR5892193","SRR5892208","SRR5892209",
"SRR5892216","SRR5892235", "SRR5892239")] <- "Non-HMP oral metagenome"
metadata$ScientificName <- factor(metadata$ScientificName) # convert back from character to factor
# add in metadata, gene type
covs$category <- metadata[as.character(covs$sample_name),"ScientificName"]
vari$category <- metadata[as.character(vari$sample_id),"ScientificName"]
covs <- unite(covs, "sample_split", c("sample_name","split_name"), remove=F)
# do the same for prevo
prevo_cov <- read.table(prevo_cov_path, header = T, sep = "\t")
prevo_var <- read.table(prevo_var_path, header = T, sep = "\t")
prevo_cov$category <- metadata[as.character(prevo_cov$sample_name),"ScientificName"]
prevo_var$category <- metadata[as.character(prevo_var$sample_id),"ScientificName"]
prevo_cov <- unite(prevo_cov, "sample_split", c("sample_name","split_name"), remove=F)
length(unique(as.character(covs$sample_name))) # this many metagenomes
## [1] 1188
Over the course of our investigations (i.e., after some preliminary plots), we noticed some outlier samples that we removed for various reasons detailed in the manuscrtipt, and so we drop them here:
prevo_cov <- prevo_cov[!(prevo_cov$sample_name == "SRR628272"),] # prevo, low quality
prevo_var <- prevo_var[!(prevo_var$sample_id == "SRR628272"),]
covs <- covs[!(covs$sample_name == "SRR628272"),] # prevotella, low quality
vari <- vari[!(vari$sample_id == "SRR628272"),]
covs <- covs[!(covs$sample_name == "SRR1779144"),] # cronobacter, infant, skews y-axis
vari <- vari[!(vari$sample_id == "SRR1779144"),]
covs <- covs[!(covs$sample_name == "SRR2047620"),] # cronobacter, stem cell kids, skews y-axis
vari <- vari[!(vari$sample_id == "SRR2047620"),]
covs <- covs[!(covs$sample_name == "SRR1038387"),] # proteus, neonatal NEC, skews y-axis
vari <- vari[!(vari$sample_id == "SRR1038387"),]
covs <- covs[!(covs$sample_name == "SRR1781983"),] # drop the high rothia sample that looks ok but skews y axis
vari <- vari[!(vari$sample_id == "SRR1781983"),]
At this point, we have 1183 metagenomes from 45 categories:
## [1] "Number of metagenomes: 1183"
## [1] "Number of sampled categories: 45"
## [1] "Homo sapiens" "metagenomes"
## [3] "metagenome" "human gut metagenome"
## [5] "gut metagenome" "sediment metagenome"
## [7] "Homo sapiens neanderthalensis" "marine metagenome"
## [9] "root" "HMP oral metagenome"
## [11] "human metagenome" "human skin metagenome"
## [13] "hydrothermal vent metagenome" "terrestrial metagenome"
## [15] "oral metagenome" "bioreactor metagenome"
## [17] "food metagenome" "human oral metagenome"
## [19] "indoor metagenome" "Non-HMP oral metagenome"
## [21] "insect metagenome" "wastewater metagenome"
## [23] "soil metagenome" "oyster metagenome"
## [25] "fossil metagenome" "pig gut metagenome"
## [27] "Gallus gallus" "bovine gut metagenome"
## [29] "Bos indicus" "freshwater metagenome"
## [31] "anaerobic digester metagenome" "hypersaline lake metagenome"
## [33] "groundwater metagenome" "microbial mat metagenome"
## [35] "milk metagenome" "upper respiratory tract metagenome"
## [37] "biogas fermenter metagenome" "compost metagenome"
## [39] "hot springs metagenome" "bovine metagenome"
## [41] "marine sediment metagenome" "soda lake metagenome"
## [43] "surface metagenome" "aquatic metagenome"
## [45] "invertebrate gut metagenome"
Filtering metagenomes based on category
The 1183 metagenomes retained still need to be filtered, as we wanted to minimize reporting spurious coverage from metagenomes not representative of that idrD sequence. In other words, we wanted to investigate only metagenomes that seem to represent the idrD sequence of interest - while one sample may contain, say, Prevotella spp. population(s) with idrD gene, that sample might not contain any Cronobacter turicensis cells, so including that sample for every sequence would just add noise.
So, we employed a filtering criterion that, for each sequence, discarded metagenomes for which more than half of the nucleotide positions in that idrD sequence were not covered at least once. Note that this is applied to each sequence separately; thus, the metagenomes retained for each sequence are not tied to each other. Here is the function we wrote to do that:
filter_mostly_zeros <- function(df, proportion_gte0=0.5) {
df_wide <- dcast(df, sample_split ~ nt_position, value.var = 'coverage')
rownames(df_wide) <- df_wide[,1]
# the !is.na part as differing gene lengths between taxa so the dcast padded lengths with na to make square
# count positions covered at least once
proportions <- apply(df_wide[,2:ncol(df_wide)], 1,
function(x) sum(1*(x[!is.na(x)] > 0))/length(x[!is.na(x)]))
keepThese <- names(proportions)[proportions >= proportion_gte0] # keep samples with enough bases covered
df <- df[as.character(df$sample_split) %in% keepThese,] # subset
return(df)
}
We also wrote a small function to bin categories together. This will be
relevant later. This function takes a dataframe (like covs) and a
named list, where each list element’s name is the desired name of the
bin to be made (e.g. 'Human oral metagenomes') and that list element
is a vector of the categories to be combined into that bin (e.g.,
c("HMP oral metagenome", "Non-HMP oral metagenome"))).
bin_categories <- function(df, bins){
df$category <- as.character(df$category)
for(b in names(bins)){
df$category[df$category %in% bins[[b]]] <- b
}
df$category <- factor(df$category, levels = unique(as.character(df$category)))
return(df)
}
With these, we generated a summary heatmap of all of the data. This is the function to generate the figure:
plot_summary_heatmap <- function(df, proportion_gte0=0.5, bins=NULL) {
if (!is.null(bins)){
df <- bin_categories(df, bins)
}
# filter non-relevant metagenomes
df <- filter_mostly_zeros(df=df, proportion_gte0 = proportion_gte0)
df$tax <- gsub("_"," ", gsub("_idrD.*$","", as.character(df$split_name)))
df$tax <- factor(df$tax, unique(as.character(df$tax)))
# summarize to get log10 mean coverage by category
df <- ddply(df, ~tax + category, summarise, LogMeanCov=log10(mean(coverage)))
ggplot(data = df, aes(x = tax, y = category)) +
geom_tile(size = 0.3, alpha = 1, aes(fill = LogMeanCov)) +
scale_fill_gradient(low = 'black', high = '#00FF00') +
xlab("idrD sequences") + ylab("Metagenome categories") + labs(fill="Log10 Mean Coverage") +
scale_x_discrete(expand=c(0,0)) + scale_y_discrete(expand=c(0,0)) + theme_minimal() +
theme(panel.grid = element_blank(), axis.line = element_line(colour = 'black', size = 0.5),
axis.text = element_text(colour='black'), axis.text.x = element_text(angle = 90, hjust = 1),
axis.ticks = element_line(colour='black', size=0.3),
panel.background = element_rect(fill = 'grey50', color = 'black'))
}
and this is how we called it to make the plot:
plot_summary_heatmap(covs)
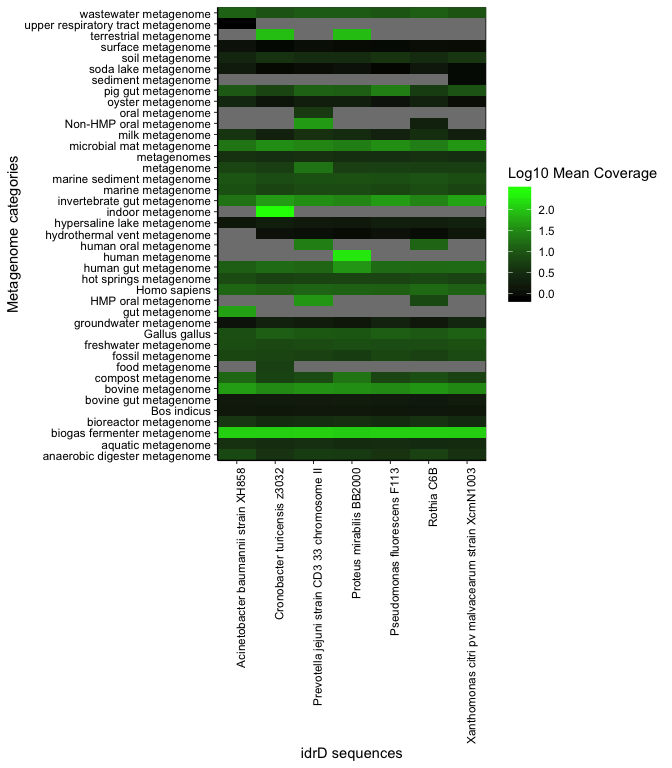
This heatmap shows, for each gene, the mean coverage of that gene for metagenomes from each category. Rows are metagenome categories, columns are idrD sequences from the different species, and each cell is shaded by the mean coverage over that entire gene from all metagenomes in that category, log10 transformed. From this plot, it becomes apparent that the various human microbiomes are interesting, and that idrD sequences from the Acinetobacter, Pseudomonas, and Xanthomonas spp. are not particularly well covered in any metagenome, except in metagenomes with evenly high coverage across all sequences (e.g. biogas fermenter metagenomes). These species’ sequences will ultimately be dropped, to focus on Proteus, Rothia, Cronobacter, and Prevotella.
Binning metagenomes by origin
Based on the heatmap, we decided to focus on human oral and human gut
metagenomes. We investigated a few different binning strageties, the one
ultimately used was bin, the other two represent bins that distinguish
HMP oral from other oral metganomes (binSplitOralHMP) and include the
FijiCOMP dataset (binWithFiji).
# metagenome bin categories
bin <- list()
bin[["aquatic"]] <- c("soda lake metagenome", "marine sediment metagenome", "marine metagenome",
"hypersaline lake metagenome", "hydrothermal vent metagenome", "hot springs metagenome",
"groundwater metagenome", "freshwater metagenome", "aquatic metagenome")
bin[["soil_probably"]] <- c("terrestrial metagenome", "surface metagenome", "soil metagenome",
"sediment metagenome")
bin[["animal"]] <- c("pig gut metagenome", "oyster metagenome", "invertebrate gut metagenome", "Gallus gallus",
"bovine metagenome", "bovine gut metagenome",
"Bos indicus", "gut metagenome")
bin[["other"]] <- c("wastewater metagenome", "upper respiratory tract metagenome", "milk metagenome",
"microbial mat metagenome", "metagenome","metagenomes", "indoor metagenome",
"Homo sapiens", "fossil metagenome", "food metagenome", "compost metagenome",
"bioreactor metagenome", "biogas fermenter metagenome", "anaerobic digester metagenome")
bin[["Non-HMP gut metagenome"]] <- c("human gut metagenome", "human metagenome")
binSplitOralHMP <- bin
bin[["Human oral metagenome"]] <- c("human oral metagenome", "HMP oral metagenome", "Non-HMP oral metagenome")
binWithFiji <- bin
binWithFiji[["Human oral metagenome"]] <- c(binWithFiji[["Human oral metagenome"]], "oral metagenome")
binSplitOralHMP[["Non-HMP oral metagenome"]] <- c("Non-HMP oral metagenome", "human oral metagenome")
binSplitOralHMP[["HMP oral metagenome"]] <- c("HMP oral metagenome")
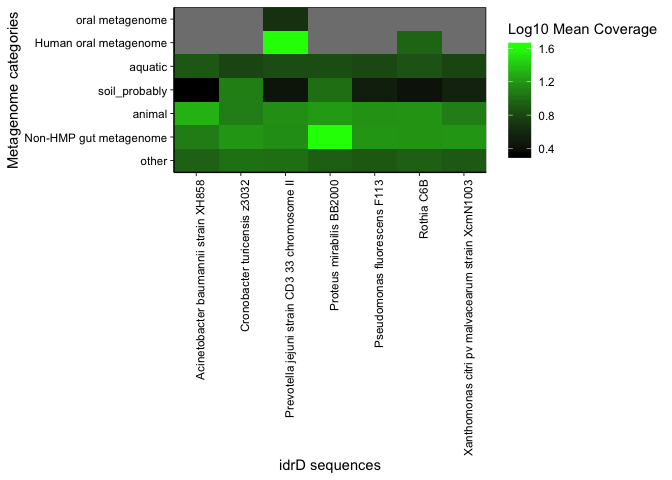
When we bin, the heatmap now looks like this, which is Supplemental Figure 3 of the paper:
plot_summary_heatmap(covs, bins = bin)
Per-nucleotide coverage to visualize region-specific coverage
Because the heatmap summarizes all nucleotide positions across many metagenomes, we wanted to understand idrD representation in metagenome populations at a finer level of detail. This also provides an opportunity to gauge idrD variability in complicated populations, which may have implications for its role in subpopulation differentiation, as suspected from its role in interstrain competition.
So, we wrote a function to plot individual metagenome coverages for each gene for each habitat we were interested in. Here is the function:
plot_mean_covs_var_combo <-function(df, v, proportion_gte0=NULL, bins=NULL, subTaxPos=NULL,
subsite=NULL, min_var_cov=0, scale_axes='free', cols = NULL,
rhsBox=NULL,rhsCols="darkgreen") {
# subset to certain metagenome categories if requested
if (!is.null(subsite)){
df <- df[df$category %in% c(subsite, unlist(bins[subsite])),]
v <- v[v$category %in% c(subsite, unlist(bins[subsite])),]
}
# make taxonomy column with pretty names
df$tax <- gsub("_"," ", gsub("_idrD.*$","", as.character(df$split_name)))
df$tax <- factor(df$tax, levels = sort(unique(as.character(df$tax))))
v$tax <- gsub("_"," ", gsub("_idrD.*$","", as.character(v$split_name)))
v$tax <- factor(v$tax, levels = sort(unique(as.character(v$tax))))
# subset to certain nuclotide windows
print("subsetting...")
if (!is.null(subTaxPos)){
df <- df[grep(paste(names(subTaxPos),collapse = "|"), as.character(df$split_name)),]
v <- v[grep(paste(names(subTaxPos),collapse = "|"), as.character(v$split_name)),]
for (subg in names(subTaxPos)){
if (is.numeric(subTaxPos[[subg]])) {
df <- filter(df, !(grepl(subg, as.character(split_name)) & (!nt_position %in% subTaxPos[[subg]])))
v <- filter(v, !(grepl(subg, as.character(split_name)) & (!pos %in% subTaxPos[[subg]])))
}
}
df$tax <- factor(df$tax, levels = sapply(names(subTaxPos), # reorder by custom value
function(x) grep(x, levels(df$tax), value = T)))
v$tax <- factor(v$tax, levels = levels(df$tax)) # reorder to match
}
print("done subsetting")
print("about to filter")
if(!is.null(proportion_gte0)){
df <- filter_mostly_zeros(df = df, proportion_gte0 = proportion_gte0)
}
v <- v[as.character(v$sample_id) %in% unique(as.character(df$sample_name)),] # keep these the same
print('done filtering')
# bin metagenome categories if needed
if (!is.null(bins)){
df <- bin_categories(df, bins)
v <- bin_categories(v, bins)
}
# count number of metagenomes to display for each sequence x metagenome combination
numMgenomes <- ddply(df, ~tax + category, summarise, nMeta=length(unique(as.character(sample_name))))
# order the metagenome categories as specified
if(!is.null(subsite)){
df$category <- factor(df$category, levels = subsite)
v$category <- factor(v$category, levels = subsite)
}
# drop any variant positions if coverage is too low (default = don't drop any variant positions)
v <- v[v$coverage >= min_var_cov,]
## re-compute the Shannon Entropy of SNPs from each position
# requires adding in counts of non-variant metagenomes and then re-calculating entropy
v <- merge(df, y =v, by.x = c('split_name', 'sample_name', 'nt_position'),
by.y = c("split_name","sample_id","pos"), all.x = T)
v <- v[!is.na(v$coverage.y),]
v$category <- v$category.x
# for positions where only some metagenomes had variance, add back the difference
v$A[v$coverage.x > v$coverage.y & v$reference == "A"] <-
v$A[v$coverage.x > v$coverage.y & v$reference == "A"] +
v$coverage.x[v$coverage.x > v$coverage.y & v$reference == "A"] -
v$coverage.y[v$coverage.x > v$coverage.y & v$reference == "A"]
v$G[v$coverage.x > v$coverage.y & v$reference == "G"] <-
v$G[v$coverage.x > v$coverage.y & v$reference == "G"] +
v$coverage.x[v$coverage.x > v$coverage.y & v$reference == "G"] -
v$coverage.y[v$coverage.x > v$coverage.y & v$reference == "G"]
v$T[v$coverage.x > v$coverage.y & v$reference == "T"] <-
v$T[v$coverage.x > v$coverage.y & v$reference == "T"] +
v$coverage.x[v$coverage.x > v$coverage.y & v$reference == "T"] -
v$coverage.y[v$coverage.x > v$coverage.y & v$reference == "T"]
v$C[v$coverage.x > v$coverage.y & v$reference == "C"] <-
v$C[v$coverage.x > v$coverage.y & v$reference == "C"] +
v$coverage.x[v$coverage.x > v$coverage.y & v$reference == "C"] -
v$coverage.y[v$coverage.x > v$coverage.y & v$reference == "C"]
# calculate the entropy for each site
variability <- ddply(v, ~nt_position + tax.x + category, summarise, origEntropy=mean(entropy),
ent = entropy.empirical(y = c(sum(A), sum(C), sum(G), sum(T)), unit = 'log2'))
# sort the taxa in the specified order
variability$tax <- factor(variability$tax.x, levels = levels(df$tax))
# coverage plot
q <- ggplot(df, aes(x = nt_position, y = coverage)) +
geom_line(size = 0.4, aes(group = sample_name, color = tax), alpha = 0.3) +
geom_text(data=numMgenomes, aes(label=nMeta), x=Inf, y = Inf, hjust = 1, vjust = 1) +
facet_grid(category~tax, scales = scale_axes, switch = "y") +
theme_minimal() + guides(color = F) +
xlab("Nucleotide position in idrD") + ylab("Metagenome coverage (X)") +
scale_x_continuous(expand=c(0,0)) + scale_y_continuous(expand=c(0,0)) +
theme(panel.grid = element_blank(), axis.line = element_line(colour = 'black', size = 0.5),
axis.text = element_text(colour='black'), axis.ticks = element_line(colour='black', size=0.3),
strip.background.y = element_rect(fill = '#dcdcdc', color = NA),
strip.background.x = element_rect(fill = '#ffffff', color=NA),
strip.text = element_text(color='black'), strip.text.x = element_text(face = 'bold'))
if (!is.null(cols)){
q <- q + scale_color_manual(values = cols)
}
# var plot
p <- ggplot(variability, aes(x = nt_position, y = ent)) +
geom_col(data = df, aes(x = nt_position, y = coverage*0), alpha = 0) +
geom_col(alpha = 1, size = 0.1, fill = 'grey20', color = NA) +
facet_grid(category~tax, scales = scale_axes) +
scale_x_continuous(expand=c(0,0)) + scale_y_continuous(expand=c(0,0), position = 'right') +
xlab("Nucleotide position in idrD") + ylab("Shannon Entropy") + theme_minimal() +
theme(panel.grid = element_blank(), axis.line = element_line(colour = 'black', size = 0.5),
axis.text = element_text(colour='black'), axis.ticks = element_line(colour='black', size=0.3),
strip.background.y = element_rect(fill = '#dcdcdc', color = NA),
strip.background.x = element_rect(fill = '#ffffff', color=NA),
strip.text = element_text(color='black'), strip.text.x = element_text(face = 'bold'))
if (!is.null(rhsBox)){
rhsBox <- data.frame(tax=names(rhsBox),matrix(unlist(rhsBox), nrow = length(rhsBox), ncol = 2, byrow = T))
rhsBox$tax_distinct <- rhsBox$tax
rhsBox$tax <- sapply(as.character(rhsBox$tax), function(x) grep(x, levels(df$tax), value = T))
rhsBox$tax <- factor(rhsBox$tax, levels = levels(df$tax))
q <- q + geom_segment(data=rhsBox,
aes(x = X1, xend = X2, y = Inf, yend = Inf), size = 1, color = rhsCols)
}
print(q)
print(p)
}
We manually defined a few additional variables to specify the taxa, sites, and regions of interest. Here they are:
# C terminal domains from taxa we care about, see ms for how start/stop positions were identified
sub4cterm <- list()
sub4cterm[["Proteus"]] <- c(4333:4746)
sub4cterm[["Rothia"]] <- c(283:702)
sub4cterm[["Cronobacter"]] <- c(4174:4572)
sub4cterm[["Prevotella"]] <- c(1816:2250)
sub4all <- list()
sub4all[["Proteus"]] <- 'all'
sub4all[["Rothia"]] <- 'all'
sub4all[["Cronobacter"]] <- 'all'
sub4all[["Prevotella"]] <- 'all'
# C terminal domains from Prevos we care about (nucleotide positions)
prevos <- list()
prevos[["jejuni"]] <- c(1816:2250)
prevos[["C561"]] <- c(238:672)
prevos[["fusca"]] <- c(835:1257)
prevos[["denticola"]] <- c(3861:4295)
# predicted Rhs start/stop sites
rhs4 <- list()
rhs4[["Proteus"]] <- c(2329,4332)
rhs4[["Rothia"]] <- c(40,276)
rhs4[["Cronobacter"]] <- c(3937,4173)
rhs4[["Prevotella"]] <- c(1585,1815)
# predicted CTD start/stop sites
rhs4[["Proteu"]] <- c(4333,4746)
rhs4[["Rothi"]] <- c(283,702)
rhs4[["Cronobacte"]] <- c(4174,4572)
rhs4[["Prevotell"]] <- c(1816,2250)
# predicted region with active fold for the 4 taxa
active4 <- list()
active4[["Proteus"]] <- c(4339,4581)
active4[["Rothia"]] <- c(286,546)
active4[["Cronobacter"]] <- c(4180,4431)
active4[["Prevotella"]] <- c(1822,2067)
# predicted region with active fold for the Prevotella
activePrevo <- list()
activePrevo[["jejuni"]] <- c(1822,2067)
activePrevo[["C561"]] <- c(238,492)
activePrevo[["fusca"]] <- c(841,1089)
# bins/sites to focus on for plotting
subsites <- c("HMP gut metagenome", "Non-HMP gut metagenome", "Human oral metagenome")#, "oral metagenome")
subsitesSplitHMP <- c(subsites, "Non-HMP oral metagenome", "HMP oral metagenome")
subsitesWithFiji <- c(subsites, "oral metagenome", "human metagenome")
subsitesWithHuman <- c(subsites, "human metagenome")
sub4cols <- c("#ee2922","#68c0d3","#aa9a4a","#8a10aa")
prevoCols <- c("#8a10aa","#ca60aa","#4a109a")
The sub* and prevos lists define the plotting windows for each taxa,
a value of all means all nucleotides will be used while a range of
numbers mean that only coverages or variability information in that
range will be plotted. Importantly, in the plot_mean_covs_var_combo
function, the filtration step happens after defining the nucleotide
range to be plotted. Thus, metagenomes are filtered based on the
proportion of non-zero-coverage positions within the nucleotide range
plotted.
The rhs4 list elements with full names (e.g. Proteus) define the
start and stop positions for each sequence’s Rhs core domain, while the
elements with truncated names (e.g., Proteu; simply to add a second
element with a searchable name) define the CTD start and stop positions.
The subsites* vectors are a convenient way to command the plotter
function to focus on a certain set of metagenome categories (including
bin names).
With this information, we can generate the plots that become Figure 4B-D.
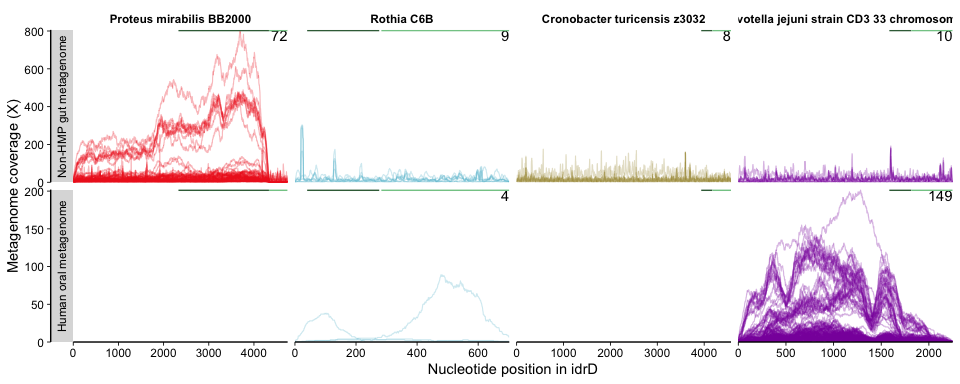
Figure 4B was generated as such:
plot_mean_covs_var_combo(df = covs, v = vari, proportion_gte0 = 0.5, bins = bin, subTaxPos = sub4all,
subsite = subsites, cols = sub4cols, rhsBox = rhs4,
rhsCols = rep(c(rep("#3a6b46",4),rep("#83ca95",4)),2))
## [1] "subsetting..."
## [1] "done subsetting"
## [1] "about to filter"
## [1] "done filtering"
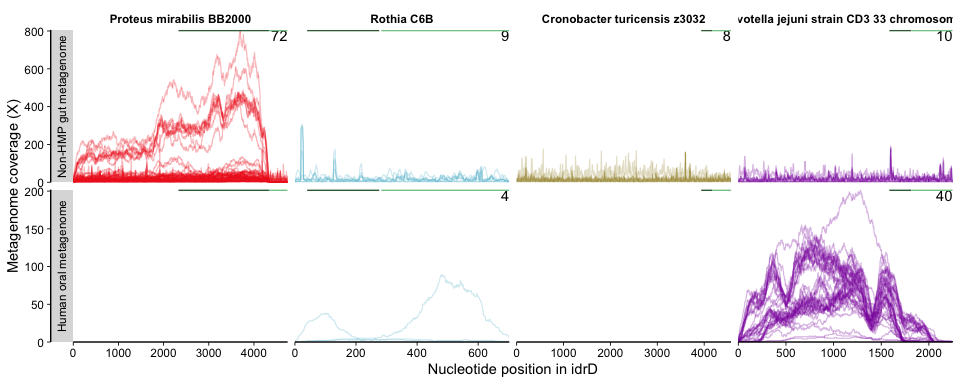
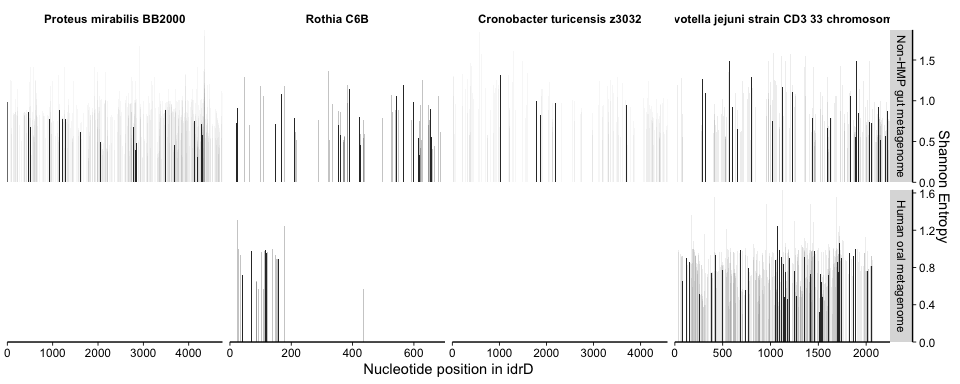
The x-axis is nucleotide position, and each subpanel represents a
different combination of metagenome category x idrD sequence. Columns of
subpanels are different species (labelled at the top), and rows are the
metagenome categories (labelled on the left). Each line traces the
coverage provided by a single metagenome, and the number of metagenomes
for that given idrD + metagenome category combination is shown in the
upper right corner of each subpanel. The second plot generated by this
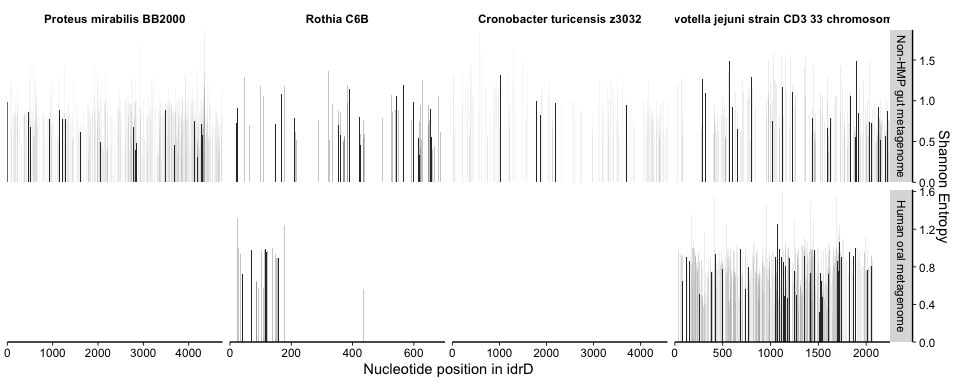
command shows the variability of each position across the different
metagenomes. The ggplot function does not like to overlay disparate y
axes, hence the separate plot. The variability is the Shannon Entropy of
all variant (and invariant) nucleotide frequencies reported by all
metagenomes for any position with variability. This is possible since
Anvi’o counted, for each metagenome, all nucleotide counts that occurred
at in a metagenome for each position with a SNP (relative to the
reference idrD sequence), which is reported in the variability file that
is vari.
It is important to note that nucleotide-level coverage patterns are always inherently wavy, besides from real biological signal, from a combination of stochastic/methodological, biological features (e.g. varying GC content). For more on this, please check out this helpful article that dives deeply into the sources of wavy coverage.
The variability information gives context into what is happening with the coverage and helps us decide whether the mapping is “real” (representing a biological population) or “spurious” (mapping artifacts or else coverage from distant populations not considered to be part of the population from which the reference sequence originated). For instance, if one region has more coverage than the rest of the genome, but also more variability, then it is a strong indicator that there is a different population (or duplication of the gene) containing that region but not the rest (or the rest is present in that population/duplicate but is so divergent that bowtie2 cannot map it). Alternatively, if in that example coverage were to go up but the variability did not, then the same multiple-population or duplication scenario exists, but that region is clearly more conserved at the nucleotide level between populations/duplicates.
To make the in-text Figure 4B, we showed only the coverage data; for the supplemental figure version, we overlaid the coverage figure on top of the variability figure with Inkscape
Note that the FijiCOMP samples were dropped, otherwise it would look like this:
plot_mean_covs_var_combo(df = covs, v = vari, proportion_gte0 = 0.5, bins = binWithFiji, subTaxPos = sub4all,
subsite = subsitesWithFiji, cols = sub4cols, rhsBox = rhs4,
rhsCols = rep(c(rep("#3a6b46",4),rep("#83ca95ff",4)),2))
## [1] "subsetting..."
## [1] "done subsetting"
## [1] "about to filter"
## [1] "done filtering"
The FijiCOMP samples (the low-coverage “noise” added to the P. jejuni x Human oral metagenome subpanel) are all very noisy and low-coverage, so potentially erroneus reads or spurious mapping, so we felt safe in discarding them.
We then focused in to just the C-terminal domain (CTD) - this is Figure
4C - by passing the function the nucleotide ranges in the CTD
sub4cterm like so:
plot_mean_covs_var_combo(df = covs, v = vari, proportion_gte0 = 0.5, bins = bin, subTaxPos = sub4cterm,
subsite = subsites, cols = sub4cols, rhsBox = active4, rhsCols = rep("#003380ff", 8))
## [1] "subsetting..."
## [1] "done subsetting"
## [1] "about to filter"
## [1] "done filtering"
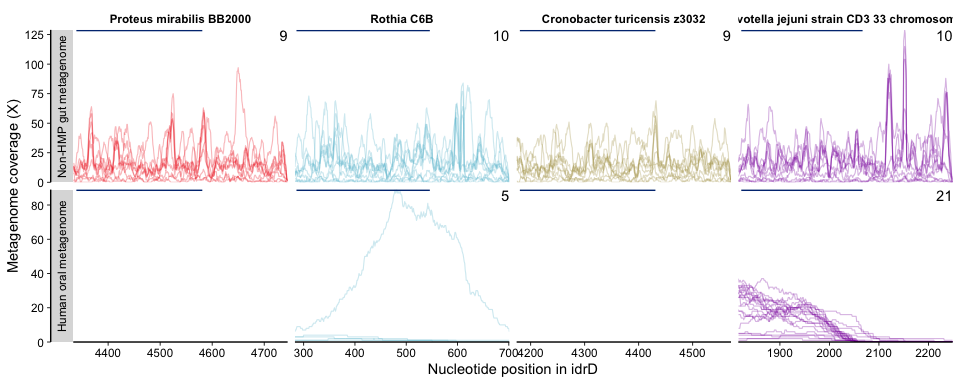
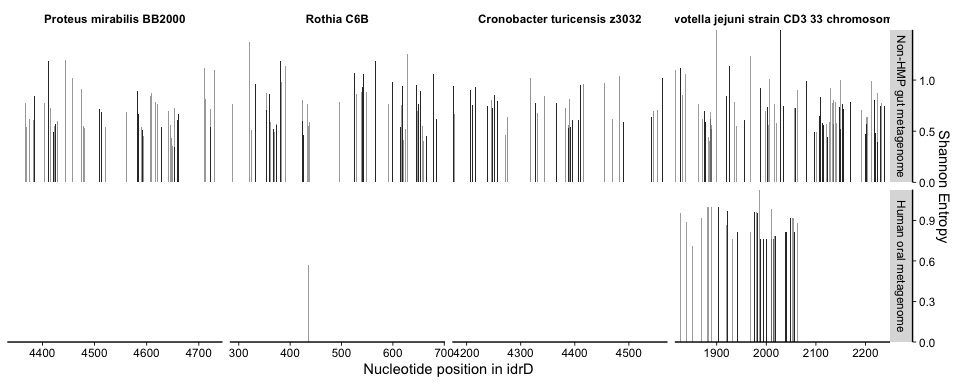
Based on this figure, we were very interested in diggng deeper into what was going on with the Prevotella in the oral cavity. The enzymatic work showed that the 5’ end of the CTD contained the active sites (region spanned by dark blue bars) while the 3’ region was more variable but still necessary (3’ truncations abolished activity). So, we were curious if the precipitous drop in metagenome coverage immediately after the first part represented a true truncation in the metagenome-sampled population(s), or whether there truly was a 3’ region(s) in the metagenome data but the P. jejuni idrD sequence was simply too distant, in nucleotide and presumably evolutionary space, to recruit/represent them.
So, we took sequences from three other Prevotella spp., mapped the same human oral metagenomes against them (using the same scripts and commands described earlier, just modified to specify the appropriate new reference files & output) - coverage data here and variability data here.
This produced the prevo_cov and prevo_var dataframes that have been
appearing in the scripts above. So then we plotted the coverage of their
CTDs like so:
plot_mean_covs_var_combo(df = prevo_cov, v = prevo_var, proportion_gte0 = 0.5, bins = bin,
subTaxPos = prevos, subsite = subsites, cols = prevoCols,
rhsBox = activePrevo, rhsCols = rep("#003380ff", 3))
## [1] "subsetting..."
## [1] "done subsetting"
## [1] "about to filter"
## [1] "done filtering"
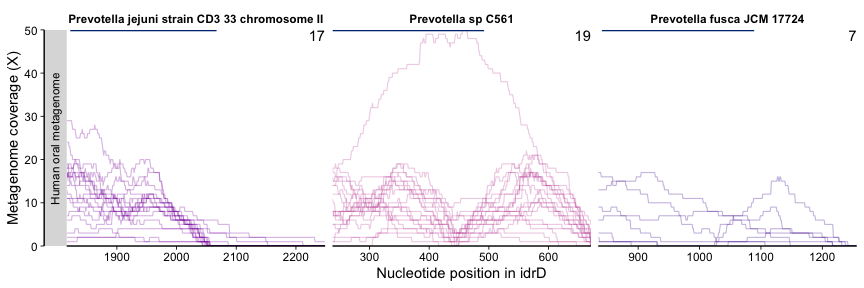
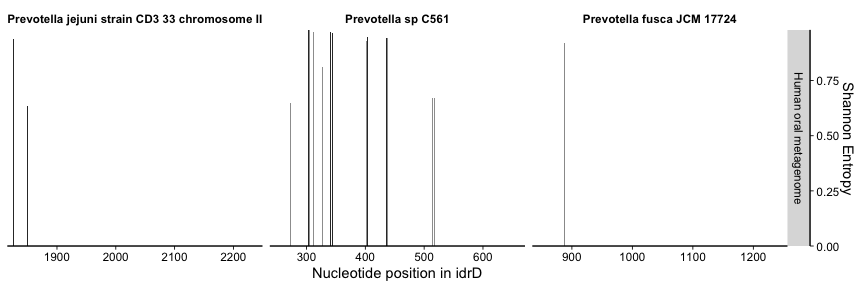
And it was immediately clear that the 3’ region of CTDs from P. sp. C561, and P. fusca to a lesser extent, recruited reads, while P. jejuni kept its original trend. It was also encouraging to see that the variability went down, reflecting that having the other Prevotella as alternative mapping targets ameliorated a significant amount of the nucleotide diversity (i.e., the P. sp. C561 sequence was more representative, and the sequence diversity was apparently due to other Prevotella populations, not some extremely unrelated population). The ‘valley’ in the middle of the P. sp. C561 CTD is puzzling, and potentially holds more insight into subpopulation diversity.